SARS-Cov-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus Or Host Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pros and Cons of Entry and Fusion Inhibitors (Review)
MOLECULAR MEDICINE REPORTS 19: 1987-1995, 2019 Investigational drugs in HIV: Pros and cons of entry and fusion inhibitors (Review) EMMANUELE VENANZI RULLO1,2, MANUELA CECCARELLI1, FABRIZIO CONDORELLI3, ALESSIO FACCIOLÀ1, GIUSEPPA VISALLI4, FRANCESCO D'ALEO1, IVANA PAOLUCCI1, BRUNO CACOPARDO5, MARILIA RITA PINZONE2-5, MICHELE DI ROSA6, GIUSEPPE NUNNARI1 and GIOVANNI F. PELLICANÒ7 1Department of Clinical and Experimental Medicine, Unit of Infectious Diseases, University of Messina, I-90124 Messina, Italy; 2Department of Pathology and Laboratory Medicine, School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA; 3Department of Pharmacological Sciences, University of Eastern Piedmont ‘A. Avogadro’, I-13100 Novara; 4Department of Biomedical and Dental Sciences and Morphofunctional Imaging, University of Messina, I-90124 Messina; 5Department of Clinical and Experimental Medicine, University of Catania, I-95123 Catania; 6Department of Biomedical and Biotechnological Sciences, Human Anatomy and Histology Section, University of Catania, I-95123 Catania; 7Department of Human Pathology of the Adult and the Developmental Age ‘G. Barresi’, Unit of Infectious Diseases, University of Messina, I-98122 Messina, Italy Received September 2, 2018; Accepted November 29, 2018 DOI: 10.3892/mmr.2019.9840 Abstract. Despite the profound changes and improve- 4. Gp41 antagonists ments reached in the field of HIV treatment, tolerability and 5. CD4 antagonists adherence to highly active antiretroviral therapy remains a 6. Discussion challenge. Furthermore, multi-experienced patients could take advantage of drugs with different mechanisms of action to combat the spread of resistance to actual therapy. For these 1. Introduction reasons identification of new HIV drugs is crucial. Among all the molecules that at present are under investigation, entry HIV continues to be an important challenge and a major and fusion inhibitors pose an interesting class owing to their global public health issue. -

Molecular Markers of Serine Protease Evolution
The EMBO Journal Vol. 20 No. 12 pp. 3036±3045, 2001 Molecular markers of serine protease evolution Maxwell M.Krem and Enrico Di Cera1 ment and specialization of the catalytic architecture should correspond to signi®cant evolutionary transitions in the Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, Box 8231, St Louis, history of protease clans. Evolutionary markers encoun- MO 63110-1093, USA tered in the sequences contributing to the catalytic apparatus would thus give an account of the history of 1Corresponding author e-mail: [email protected] an enzyme family or clan and provide for comparative analysis with other families and clans. Therefore, the use The evolutionary history of serine proteases can be of sequence markers associated with active site structure accounted for by highly conserved amino acids that generates a model for protease evolution with broad form crucial structural and chemical elements of applicability and potential for extension to other classes of the catalytic apparatus. These residues display non- enzymes. random dichotomies in either amino acid choice or The ®rst report of a sequence marker associated with serine codon usage and serve as discrete markers for active site chemistry was the observation that both AGY tracking changes in the active site environment and and TCN codons were used to encode active site serines in supporting structures. These markers categorize a variety of enzyme families (Brenner, 1988). Since serine proteases of the chymotrypsin-like, subtilisin- AGY®TCN interconversion is an uncommon event, it like and a/b-hydrolase fold clans according to phylo- was reasoned that enzymes within the same family genetic lineages, and indicate the relative ages and utilizing different active site codons belonged to different order of appearance of those lineages. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Identification of New Substrates and Physiological Relevance
Université de Montréal The Multifaceted Proprotein Convertases PC7 and Furin: Identification of New Substrates and Physiological Relevance Par Stéphanie Duval Biologie Moléculaire, Faculté de médecine Thèse présentée en vue de l’obtention du grade de Philosophiae doctor (Ph.D) en Biologie moléculaire, option médecine cellulaire et moléculaire Avril 2020 © Stéphanie Duval, 2020 Résumé Les proprotéines convertases (PCs) sont responsables de la maturation de plusieurs protéines précurseurs et sont impliquées dans divers processus biologiques importants. Durant les 30 dernières années, plusieurs études sur les PCs se sont traduites en succès cliniques, toutefois les fonctions spécifiques de PC7 demeurent obscures. Afin de comprendre PC7 et d’identifier de nouveaux substrats, nous avons généré une analyse protéomique des protéines sécrétées dans les cellules HuH7. Cette analyse nous a permis d’identifier deux protéines transmembranaires de fonctions inconnues: CASC4 et GPP130/GOLIM4. Au cours de cette thèse, nous nous sommes aussi intéressé au rôle de PC7 dans les troubles comportementaux, grâce à un substrat connu, BDNF. Dans le chapitre premier, je présenterai une revue de la littérature portant entre autres sur les PCs. Dans le chapitre II, l’étude de CASC4 nous a permis de démontrer que cette protéine est clivée au site KR66↓NS par PC7 et Furin dans des compartiments cellulaires acides. Comme CASC4 a été rapporté dans des études de cancer du sein, nous avons généré des cellules MDA- MB-231 exprimant CASC4 de type sauvage et avons démontré une diminution significative de la migration et de l’invasion cellulaire. Ce phénotype est causé notamment par une augmentation du nombre de complexes d’adhésion focale et peut être contrecarré par la surexpression d’une protéine CASC4 mutante ayant un site de clivage optimale par PC7/Furin ou encore en exprimant une protéine contenant uniquement le domaine clivé N-terminal. -

(KPIC) PPO and Out-Of- Area Indemnity (OOA) Drug Formulary with Specialty Drug Tier
Kaiser Permanente Insurance Company (KPIC) PPO and Out-of- Area Indemnity (OOA) Drug Formulary with Specialty Drug Tier This Drug Formulary was updated: September 1, 2021 NOTE: This drug formulary is updated often and is subject to change. Upon revision, all previous versions of the drug formulary are no longer in effect. This document contains information regarding the drugs that are covered when you participate in the California Nongrandfathered PPO and Out-of- Area Indemnity (OOA) Health Insurance Plans with specialty drug tier offered by Kaiser Permanente Insurance Company (KPIC) and fill your prescription at a MedImpact network pharmacy. Access to the most current version of the Formulary can be obtained by visiting kp.org/kpic-ca-rx-ppo-ngf. For help understanding your KPIC insurance plan benefits, including cost sharing for drugs under the prescription drug benefit and under the medical benefit, please call 1-800-788-0710 or 711 (TTY) Monday through Friday, 7a.m. to 7p.m. For help with this Formulary, including the processes for submitting an exception request and requesting prior authorization and step therapy exceptions, please call MedImpact 24 hours a day, 7 days a week, at 1-800-788-2949 or 711 (TTY). For cost sharing information for the outpatient prescription drug benefits in your specific plan, please visit: kp.org/kpic-ca-rx-ppo-ngf. For help in your preferred language, please see the Kaiser Permanente Insurance Company Notice of Language Assistance in this document. KPIC PPO NGF Table of Contents Informational Section................................................................................................................................2 -

Cell Surface–Anchored Serine Proteases in Cancer Progression and Metastasis
Cancer and Metastasis Reviews (2019) 38:357–387 https://doi.org/10.1007/s10555-019-09811-7 Cell surface–anchored serine proteases in cancer progression and metastasis Carly E. Martin1,2 & Karin List1,2 Published online: 16 September 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Over the last two decades, a novel subgroup of serine proteases, the cell surface–anchored serine proteases, has emerged as an important component of the human degradome, and several members have garnered significant attention for their roles in cancer progression and metastasis. A large body of literature describes that cell surface–anchored serine proteases are deregulated in cancer and that they contribute to both tumor formation and metastasis through diverse molecular mechanisms. The loss of precise regulation of cell surface–anchored serine protease expression and/or catalytic activity may be contributing to the etiology of several cancer types. There is therefore a strong impetus to understand the events that lead to deregulation at the gene and protein levels, how these precipitate in various stages of tumorigenesis, and whether targeting of selected proteases can lead to novel cancer intervention strategies. This review summarizes current knowledge about cell surface–anchored serine proteases and their role in cancer based on biochemical characterization, cell culture–based studies, expression studies, and in vivo experiments. Efforts to develop inhibitors to target cell surface–anchored serine proteases in cancer therapy will also be summarized. Keywords Type II transmembrane serine proteases . Cancer . Matriptase . Hepsin . TMPRSS2 . TMPRSS3 . TMPRSS4 . Prostasin . Testisin 1 Introduction PRSS31, transmembrane tryptase, and transmembrane prote- ase γ1) is expressed in cells of hematopoietic origin and has The class of serine proteases contains 175 predicted members been studied most extensively in mast cells [2]. -
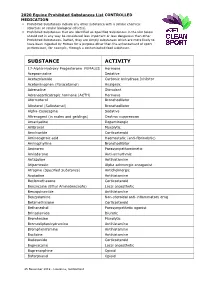
2020 Equine Prohibited Substances List CONTROLLED MEDICATION
2020 Equine Prohibited Substances List CONTROLLED MEDICATION . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). Prohibited Substances that are identified as Specified Substances in the List below should not in any way be considered less important or less dangerous than other Prohibited Substances. Rather, they are simply substances which are more likely to have been ingested by Horses for a purpose other than the enhancement of sport performance, for example, through a contaminated food substance. SUBSTANCE ACTIVITY 17-Alpha-Hydroxy Progesterone FEMALES Hormone Acepromazine Sedative Acetazolamide Carbonic Anhydrase Inhibitor Acetominophen (Paracetamol) Analgesic Adrenaline Stimulant Adrenocorticotropic hormone (ACTH) Hormone Aformoterol Bronchodilator Albuterol (Salbutamol) Bronchodilator Alpha-Casozepine Sedative Altrenogest (in males and geldings) Oestrus suppression Amantadine Dopaminergic Ambroxol Mucolytic Amcinonide Corticosteroid Aminocaproic acid Haemostatic (anti-fibrinolytic) Aminophylline Bronchodilator Aminorex Parasympathomimetic Amiodarone Anti-arrhythmic Antazoline Antihistamine Atipamezole Alpha adrenergic antagonist Atropine (Specified Substance) Anticholinergic Azatadine Antihistamine Beclomethasone Corticosteroid Benzocaine (Ethyl Aminobenzoate) Local anaesthetic Benzquinamide Antihistamine Benzydamine Non-steroidal anti-inflammatory drug Betamethasone Corticosteroid Bethanechol Parasympathetic agonist Brinzolamide Diuretic Bromhexine Mucolytic Bromodiphenhydramine -

National OTC Medicines List
National OTC Medicines List ‐ DraŌ 01 DRAFT National OTC Medicines List Draft 01 Ministry of Public Health of Lebanon This list was prepared under the guidance of His Excellency Minister Waêl Abou Faour andDRAFT the supervision of the Director General Dr. Walid Ammar. Editors Rita KARAM, Pharm D. PhD. Myriam WATFA, Pharm D Ghassan HAMADEH, MD.CPE FOREWORD According to the French National Agency for Medicines and Health Products Safety (ANSM), Over-the-counter (OTC) drugs are medicines that are accessible to patients in pharmacies, based on criteria set to safeguard patients’ safety. Due to their therapeutic class, these medicines could be dispensed without physician’s intervention for diagnostic, treatment initiation or maintenance purposes. Moreover, their dosage, treatment period and Package Insert Leaflet should be suitable for OTC classification. The packaging size should be in accordance with the dosage and treatment period. According to ArticleDRAFT 43 of the Law No.367 issued in 1994 related to the pharmacy practice, and the amendment of Articles 46 and 47 by Law No.91 issued in 2010, pharmacists do not have the right to dispense any medicine that is not requested by a unified prescription, unless the medicine is mentioned in a list which is established by pharmacists and physicians’ syndicates. In this regard, the Ministry of Public Health (MoPH) developed the National OTC Medicines List, and presentedit in a scientific, objective, reliable, and accessible listing. The OTC List was developed by a team of pharmacists and physicians from the Ministry of Public Health (MoPH). In order to ensure a safe and effective self- medicationat the pharmacy level, several pharmaceutical categories (e.g. -

PRESS RELEASE IAS 2019: Viiv Healthcare Showcasing Innovation in HIV Science
NOTE TO EDITORS: ViiV Healthcare will hold a press conference at IAS 2019 on Tuesday 23 July, 9:00 – 9:45 am CDT to preview abstracts to be presented during the conference. To register, please visit: http://bit.ly/IAS-ViiV PRESS RELEASE IAS 2019: ViiV Healthcare showcasing innovation in HIV science Data to be presented span the company’s diverse portfolio, challenging the current treatment paradigm and investigating new options to meet the evolving needs of people living with HIV London, 15 July 2019 – ViiV Healthcare, the global specialist HIV company majority-owned by GSK, with Pfizer Inc. and Shionogi Limited as shareholders, announced today that 20 abstracts from its portfolio of late-stage pipeline and authorised HIV treatments will be presented at the 10th International AIDS Society Conference on HIV Science (IAS 2019) in Mexico City, 21-24 July, in Mexico City, Mexico. Since its inception 10 years ago, as the only pharmaceutical company solely focused on HIV, ViiV Healthcare continues its industry-leading commitment by delivering scientific advances that address the needs of the HIV community. The data presented at IAS 2019 further build upon the company’s innovative approach to research and development by investigating treatments that have the potential to reduce the number of medicines people living with HIV take during their lifetime and provide a range of options that meet their diverse and evolving needs. Highlights at IAS 2019 include the presentation of safety and efficacy data for the 2-drug regimen (2DR) of dolutegravir plus lamivudine in treatment-naïve and treatment-experienced patients; longer- term clinical trial data of investigational fostemsavir in heavily treatment-experienced patients; and pooled clinical trial data and patient-reported outcomes from the investigational long-acting, injectable 2DR of cabotegravir plus rilpivirine. -

MOLECULAR MODELING STUDIES on HIV-1 REVERSE TRANSCRIPTASE (RT) and HEAT SHOCK PROTEIN (Hsp) 90 AS a POTENTIAL ANTI-HIV-1 TARGET
MOLECULAR MODELING STUDIES ON HIV-1 REVERSE TRANSCRIPTASE (RT) AND HEAT SHOCK PROTEIN (Hsp) 90 AS A POTENTIAL ANTI-HIV-1 TARGET FAVOURITE NONTANDO CELE (BSc.) A thesis submitted to the College of Health Sciences, University of KwaZulu-Natal, Westville, in fulfillment of the requirements of the degree of Master of Medical Sciences Supervisor Professor Mahmoud Soliman Durban 2015 MOLECULAR MODELING STUDIES ON HIV-1 REVERSE TRANSCRIPTASE (RT) AND HEAT SHOCK PROTEIN (Hsp) 90 AS A POTENTIAL ANTI-HIV-1 TARGET FAVOURITE NONTANDO CELE A thesis submitted to the School of Health Science, University of KwaZulu-Natal, Westville Campus, for the degree of Master of Medical Science in Pharmaceutical Chemistry. This is the thesis in which the chapters are written as a set of discrete research publications, with an overall introduction and final summary. This is to certify that the contents of this thesis are the original research work of Miss Favourite Nontando Cele. As the candidate’s supervisor, I have approved this thesis for submission. Supervisor: Signed: ----------- Name: Prof. Mahmoud E. Soliman Date: ---------------------- ii ABSTRACT Human immunodeficiency virus (HIV) infection is the leading cause of death globally. This dissertation addresses two HIV-1 target proteins namely, HIV-1 Reverse Transcriptase (RT) and Heat shock protein (Hsp) 90. More specifically for HIV-1 RT, a case study for the identification of potential inhibitors as anti-HIV agents was carried out. A more refined virtual screening (VS) approach was implemented, which was an improvement on work previously published by our group- “target-bound pharmacophore modeling approach”. This study generated a pharmacophore library based only on highly contributing amino acid residues (HCAAR), instead of arbitrary pharmacophores, most commonly used in the conventional approaches in literature. -

Guidelines for the Use of Antiretroviral Agents in Adults and Adolescent Living With
Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV Developed by the DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents – A Working Group of the Office of AIDS Research Advisory Council (OARAC) How to Cite the Adult and Adolescent Guidelines: Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services. Available at https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/ AdultandAdolescentGL.pdf. Accessed [insert date] [insert page number, table number, etc. if applicable] It is emphasized that concepts relevant to HIV management evolve rapidly. The Panel has a mechanism to update recommendations on a regular basis, and the most recent information is available on the HIVinfo Web site (http://hivinfo.nih.gov). What’s New in the Guidelines? August 16, 2021 Hepatitis C Virus/HIV Coinfection • Table 18 of this section has been updated to include recommendations regarding concomitant use of fostemsavir or long acting cabotegravir plus rilpivirine with different hepatitis C treatment regimens. June 3, 2021 What to Start • Since the release of the last guidelines, updated data from the Botswana Tsepamo study have shown that the prevalence of neural tube defects (NTD) associated with dolutegravir (DTG) use during conception is much lower than previously reported. Based on these new data, the Panel now recommends that a DTG-based regimen can be prescribed for most people with HIV who are of childbearing potential. Before initiating a DTG-based regimen, clinicians should discuss the risks and benefits of using DTG with persons of childbearing potential, to allow them to make an informed decision. -

Mucoactive Agents for Airway Mucus Hypersecretory Diseases
Mucoactive Agents for Airway Mucus Hypersecretory Diseases Duncan F Rogers PhD FIBiol Introduction Sputum Profile of Airway Inflammation and Mucus Hypersecretory Phenotype in Asthma, COPD, and CF Which Aspect of Airway Mucus Hypersecretion to Target? Theoretical Requirements for Effective Therapy of Airway Mucus Hypersecretion Current Recommendations for Clinical Use of Mucolytic Drugs Mucoactive Drugs N-Acetylcysteine: How Does it Work? Does it Work? Dornase Alfa Hypertonic Saline Surfactant Analysis Summary Airway mucus hypersecretion is a feature of a number of severe respiratory diseases, including asthma, chronic obstructive pulmonary disease (COPD), and cystic fibrosis (CF). However, each disease has a different airway inflammatory response, with consequent, and presumably linked, mucus hypersecretory phenotype. Thus, it is possible that optimal treatment of the mucus hyper- secretory element of each disease should be disease-specific. Nevertheless, mucoactive drugs are a longstanding and popular therapeutic option, and numerous compounds (eg, N-acetylcysteine, erdosteine, and ambroxol) are available for clinical use worldwide. However, rational recommen- dation of these drugs in guidelines for management of asthma, COPD, or CF has been hampered by lack of information from well-designed clinical trials. In addition, the mechanism of action of most of these drugs is unknown. Consequently, although it is possible to categorize them according to putative mechanisms of action, as expectorants (aid and/or induce cough), mucolytics (thin