Abstract Algorithms for the Alignment and Visualization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Next Generation Web Scanning Presentation
Next generation web scanning New Zealand: A case study First presented at KIWICON III 2009 By Andrew Horton aka urbanadventurer NZ Web Recon Goal: To scan all of New Zealand's web-space to see what's there. Requirements: – Targets – Scanning – Analysis Sounds easy, right? urbanadventurer (Andrew Horton) www.morningstarsecurity.com Targets urbanadventurer (Andrew Horton) www.morningstarsecurity.com Targets What does 'NZ web-space' mean? It could mean: •Geographically within NZ regardless of the TLD •The .nz TLD hosted anywhere •All of the above For this scan it means, IPs geographically within NZ urbanadventurer (Andrew Horton) www.morningstarsecurity.com Finding Targets We need creative methods to find targets urbanadventurer (Andrew Horton) www.morningstarsecurity.com DNS Zone Transfer urbanadventurer (Andrew Horton) www.morningstarsecurity.com Find IP addresses on IRC and by resolving lots of NZ websites 58.*.*.* 60.*.*.* 65.*.*.* 91.*.*.* 110.*.*.* 111.*.*.* 113.*.*.* 114.*.*.* 115.*.*.* 116.*.*.* 117.*.*.* 118.*.*.* 119.*.*.* 120.*.*.* 121.*.*.* 122.*.*.* 123.*.*.* 124.*.*.* 125.*.*.* 130.*.*.* 131.*.*.* 132.*.*.* 138.*.*.* 139.*.*.* 143.*.*.* 144.*.*.* 146.*.*.* 150.*.*.* 153.*.*.* 156.*.*.* 161.*.*.* 162.*.*.* 163.*.*.* 165.*.*.* 166.*.*.* 167.*.*.* 192.*.*.* 198.*.*.* 202.*.*.* 203.*.*.* 210.*.*.* 218.*.*.* 219.*.*.* 222.*.*.* 729,580,500 IPs. More than we want to try. urbanadventurer (Andrew Horton) www.morningstarsecurity.com IP address blocks in the IANA IPv4 Address Space Registry Prefix Designation Date Whois Status [1] ----- -

Pipenightdreams Osgcal-Doc Mumudvb Mpg123-Alsa Tbb
pipenightdreams osgcal-doc mumudvb mpg123-alsa tbb-examples libgammu4-dbg gcc-4.1-doc snort-rules-default davical cutmp3 libevolution5.0-cil aspell-am python-gobject-doc openoffice.org-l10n-mn libc6-xen xserver-xorg trophy-data t38modem pioneers-console libnb-platform10-java libgtkglext1-ruby libboost-wave1.39-dev drgenius bfbtester libchromexvmcpro1 isdnutils-xtools ubuntuone-client openoffice.org2-math openoffice.org-l10n-lt lsb-cxx-ia32 kdeartwork-emoticons-kde4 wmpuzzle trafshow python-plplot lx-gdb link-monitor-applet libscm-dev liblog-agent-logger-perl libccrtp-doc libclass-throwable-perl kde-i18n-csb jack-jconv hamradio-menus coinor-libvol-doc msx-emulator bitbake nabi language-pack-gnome-zh libpaperg popularity-contest xracer-tools xfont-nexus opendrim-lmp-baseserver libvorbisfile-ruby liblinebreak-doc libgfcui-2.0-0c2a-dbg libblacs-mpi-dev dict-freedict-spa-eng blender-ogrexml aspell-da x11-apps openoffice.org-l10n-lv openoffice.org-l10n-nl pnmtopng libodbcinstq1 libhsqldb-java-doc libmono-addins-gui0.2-cil sg3-utils linux-backports-modules-alsa-2.6.31-19-generic yorick-yeti-gsl python-pymssql plasma-widget-cpuload mcpp gpsim-lcd cl-csv libhtml-clean-perl asterisk-dbg apt-dater-dbg libgnome-mag1-dev language-pack-gnome-yo python-crypto svn-autoreleasedeb sugar-terminal-activity mii-diag maria-doc libplexus-component-api-java-doc libhugs-hgl-bundled libchipcard-libgwenhywfar47-plugins libghc6-random-dev freefem3d ezmlm cakephp-scripts aspell-ar ara-byte not+sparc openoffice.org-l10n-nn linux-backports-modules-karmic-generic-pae -
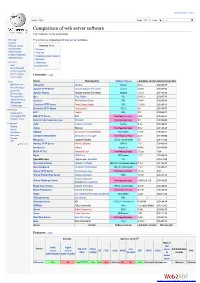
Comparison of Web Server Software from Wikipedia, the Free Encyclopedia
Create account Log in Article Talk Read Edit ViewM ohrisetory Search Comparison of web server software From Wikipedia, the free encyclopedia Main page This article is a comparison of web server software. Contents Featured content Contents [hide] Current events 1 Overview Random article 2 Features Donate to Wikipedia 3 Operating system support Wikimedia Shop 4 See also Interaction 5 References Help 6 External links About Wikipedia Community portal Recent changes Overview [edit] Contact page Tools Server Developed by Software license Last stable version Latest release date What links here AOLserver NaviSoft Mozilla 4.5.2 2012-09-19 Related changes Apache HTTP Server Apache Software Foundation Apache 2.4.10 2014-07-21 Upload file Special pages Apache Tomcat Apache Software Foundation Apache 7.0.53 2014-03-30 Permanent link Boa Paul Phillips GPL 0.94.13 2002-07-30 Page information Caudium The Caudium Group GPL 1.4.18 2012-02-24 Wikidata item Cite this page Cherokee HTTP Server Álvaro López Ortega GPL 1.2.103 2013-04-21 Hiawatha HTTP Server Hugo Leisink GPLv2 9.6 2014-06-01 Print/export Create a book HFS Rejetto GPL 2.2f 2009-02-17 Download as PDF IBM HTTP Server IBM Non-free proprietary 8.5.5 2013-06-14 Printable version Internet Information Services Microsoft Non-free proprietary 8.5 2013-09-09 Languages Jetty Eclipse Foundation Apache 9.1.4 2014-04-01 Čeština Jexus Bing Liu Non-free proprietary 5.5.2 2014-04-27 Galego Nederlands lighttpd Jan Kneschke (Incremental) BSD variant 1.4.35 2014-03-12 Português LiteSpeed Web Server LiteSpeed Technologies Non-free proprietary 4.2.3 2013-05-22 Русский Mongoose Cesanta Software GPLv2 / commercial 5.5 2014-10-28 中文 Edit links Monkey HTTP Server Monkey Software LGPLv2 1.5.1 2014-06-10 NaviServer Various Mozilla 1.1 4.99.6 2014-06-29 NCSA HTTPd Robert McCool Non-free proprietary 1.5.2a 1996 Nginx NGINX, Inc. -

'A Draft Sequence of the Neandertal Genome'
A Draft Sequence of the Neandertal Genome Richard E. Green, et al. Science 328, 710 (2010); DOI: 10.1126/science.1188021 This copy is for your personal, non-commercial use only. If you wish to distribute this article to others, you can order high-quality copies for your colleagues, clients, or customers by clicking here. Permission to republish or repurpose articles or portions of articles can be obtained by following the guidelines here. The following resources related to this article are available online at www.sciencemag.org (this information is current as of May 7, 2010 ): Updated information and services, including high-resolution figures, can be found in the online version of this article at: http://www.sciencemag.org/cgi/content/full/328/5979/710 Supporting Online Material can be found at: http://www.sciencemag.org/cgi/content/full/328/5979/710/DC1 This article cites 81 articles, 29 of which can be accessed for free: http://www.sciencemag.org/cgi/content/full/328/5979/710#otherarticles on May 7, 2010 This article has been cited by 1 articles hosted by HighWire Press; see: http://www.sciencemag.org/cgi/content/full/328/5979/710#otherarticles This article appears in the following subject collections: Immunology http://www.sciencemag.org/cgi/collection/immunology www.sciencemag.org Downloaded from Science (print ISSN 0036-8075; online ISSN 1095-9203) is published weekly, except the last week in December, by the American Association for the Advancement of Science, 1200 New York Avenue NW, Washington, DC 20005. Copyright 2010 by the American Association for the Advancement of Science; all rights reserved. -

Delivering Web Content
Delivering web content GET /rfc.html HTTP/1.1 Host: www.ie.org GET /rfc.html HTTP/1.1 User-agent: Mozilla/4.0 Host: www.ie.org GET /rfc.html HTTP/1.1 User-agent: Mozilla/4.0 Host: www.ie.org User-agent: Mozilla/4.0 GET /rfc.html HTTP/1.1 Host: www.ie.org GET /rfc.html HTTP/1.1 User-agent: Mozilla/4.0 Host: www.ie.org User-agent: Mozilla/4.0 CSCI 470: Web Science • Keith Vertanen • Copyright © 2011 Overview • HTTP protocol review – Request and response format – GET versus POST • Stac and dynamic content – Client-side scripIng – Server-side extensions • CGI • Server-side includes • Server-side scripng • Server modules • … 2 HTTP protocol • HyperText Transfer Protocol (HTTP) – Simple request-response protocol – Runs over TCP, port 80 – ASCII format request and response headers GET /rfc.html HTTP/1.1 Host: www.ie.org Method User-agent: Mozilla/4.0 Header lines Carriage return, line feed indicates end of request 3 TCP details MulIple Persistent Persistent connecIons and connecIon and connecIon and sequenIal requests. sequenIal requests. pipelined requests. 4 HTTP request GET /rfc.html HTTP/1.1 Host: www.ie7.org User-agent: Mozilla/4.0 POST /login.html HTTP/1.1 Host: www.store.com User-agent: Mozilla/4.0 Content-Length: 27 Content-Type: applicaon/x-www-form-urlencoded userid=joe&password=guessme 5 HTTP response • Response from server – Status line: protocol version, status code, status phrase – Response headers: extra info – Body: opIonal data HTTP/1.1 200 OK Date: Thu, 17 Nov 2011 15:54:10 GMT Server: Apache/2.2.16 (Debian) Last-Modified: Wed, -

The Weblate Manual Versão 4.2.1 Michal Čihař
The Weblate Manual Versão 4.2.1 Michal Čihař 21 ago., 2020 Documentação de utilizador 1 Documentação de utilizador 1 1.1 Básico do Weblate .......................................... 1 1.2 Registo e perfil de utilizador ..................................... 1 1.3 Traduzir usando o Weblate ...................................... 10 1.4 Descarregar e enviar traduções .................................... 20 1.5 Verificações e correções ....................................... 22 1.6 Searching ............................................... 38 1.7 Application developer guide ..................................... 42 1.8 Fluxos de trabalho de tradução .................................... 62 1.9 Frequently Asked Questions ..................................... 66 1.10 Formatos de ficheiros suportados ................................... 73 1.11 Integração de controlo de versões ................................... 91 1.12 Weblate’s REST API ......................................... 98 1.13 Cliente Weblate ............................................ 137 1.14 Weblate’s Python API ........................................ 141 2 Documentação de administrador 143 2.1 Configuration instructions ....................................... 143 2.2 Weblate deployments ......................................... 195 2.3 Upgrading Weblate .......................................... 196 2.4 Fazer backup e mover o Weblate ................................... 200 2.5 Autenticação ............................................. 205 2.6 Controlo de acesso ......................................... -

The Buildroot User Manual I
The Buildroot user manual i The Buildroot user manual The Buildroot user manual ii Contents I Getting started 1 1 About Buildroot 2 2 System requirements 3 2.1 Mandatory packages.................................................3 2.2 Optional packages...................................................4 3 Getting Buildroot 5 4 Buildroot quick start 6 5 Community resources 8 II User guide 9 6 Buildroot configuration 10 6.1 Cross-compilation toolchain............................................. 10 6.1.1 Internal toolchain backend.......................................... 11 6.1.2 External toolchain backend.......................................... 11 6.1.2.1 External toolchain wrapper.................................... 12 6.2 /dev management................................................... 13 6.3 init system....................................................... 13 7 Configuration of other components 15 8 General Buildroot usage 16 8.1 make tips....................................................... 16 8.2 Understanding when a full rebuild is necessary................................... 17 8.3 Understanding how to rebuild packages....................................... 18 8.4 Offline builds..................................................... 18 8.5 Building out-of-tree.................................................. 18 The Buildroot user manual iii 8.6 Environment variables................................................ 19 8.7 Dealing efficiently with filesystem images...................................... 19 8.8 Graphing the dependencies -

Python Guide Documentation Release 0.0.1
Python Guide Documentation Release 0.0.1 Kenneth Reitz fev 10, 2019 Sumário 1 Começando com Python 3 1.1 Picking a Python Interpreter (3 vs 2)...................................3 1.2 Instalando Python corretamente......................................6 1.3 Instalando Pyhton 3 no Mac OS X....................................7 1.4 Installing Python 3 on Windows..................................... 10 1.5 Installing Python 3 on Linux....................................... 11 1.6 Installing Python 2 on Mac OS X.................................... 13 1.7 Installing Python 2 on Windows..................................... 15 1.8 Installing Python 2 on Linux....................................... 17 1.9 Pipenv & Virtual Environments..................................... 18 1.10 Lower level: virtualenv.......................................... 21 2 Ambientes de desenvolvimento em Python 25 2.1 Seu ambiente de desenvolvimento.................................... 26 2.2 Further Configuration of pip and Virtualenv............................... 32 3 Escrevendo Ótimos códigos em Python 35 3.1 Estruturando seu projeto......................................... 35 3.2 Estilo de código............................................. 47 3.3 Lendo Ótimos Códigos.......................................... 59 3.4 Documentação.............................................. 60 3.5 Testando seu código........................................... 64 3.6 Logging.................................................. 69 3.7 Common Gotchas........................................... -

Migration from Windows to Linux for a Small Engineering Firm "A&G Associates"
Rochester Institute of Technology RIT Scholar Works Theses 2004 Migration from Windows to Linux for a small engineering firm "A&G Associates" Trimbak Vohra Follow this and additional works at: https://scholarworks.rit.edu/theses Recommended Citation Vohra, Trimbak, "Migration from Windows to Linux for a small engineering firm A&G" Associates"" (2004). Thesis. Rochester Institute of Technology. Accessed from This Thesis is brought to you for free and open access by RIT Scholar Works. It has been accepted for inclusion in Theses by an authorized administrator of RIT Scholar Works. For more information, please contact [email protected]. Migration from Windows to Linux for a Small Engineering Firm "A&G Associates" (H ' _T ^^L. WBBmBmBBBBmb- Windows Linux by Trimbak Vohra Thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Information Technology Rochester Institute of Technology B. Thomas Golisano College of Computing and Information Sciences Date: December 2, 2004 12/B2/28B2 14:46 5854752181 RIT INFORMATION TECH PAGE 02 Rochester Institute of Teehnology B. Thomas Golisano College of Computing and Information Sciences Master of Science in Information Technology Thesis Approval Form Student Name: Trimbak Vohra Thesis Title: Migration from Windows to Unux for a Small Engineeriog Firm "A&G Associates" Thesis Committee Name Signature Date Luther Troell luther IrQell, Ph.D ttL ",j7/Uy Chair G. L. Barido Prof. ~~orge Barido ? - Dec:. -cl7' Committee Member Thomas Oxford Mr. Thomas OxfocQ \ 2. L~( Q~ Committee Member Thesis Reproduction Permission Form Rochester Institute of Technology B. Thomas Golisano College of Computing and Information Sciences Master of Science in Information Technology Migration from Windows to Linux for a Small Engineering Firm "A&G Associates" I,Trimbak Vohra, hereby grant permission to the Wallace Library of the Rochester Institute of Technology to reproduce my thesis in whole or in part. -

Testing Security for Internet of Things a Survey on Vulnerabilities in IP Cameras
Testing Security for Internet of Things A Survey on Vulnerabilities in IP Cameras Kim Jonatan Wessel Bjørneset Master’s Thesis Autumn 2017 Testing Security for Internet of Things Kim Jonatan Wessel Bjørneset 23rd November 2017 Abstract The number of devices connected to the Internet is growing rapidly. Many of these devices are referred to as IoT-devices. These are easy to connect and access over the Internet. Many of these, though, come with security flaws and vulnerabilities which make them easy targets for attackers. This is something that has been reviewed a lot in media lately. An IP camera is a typical example of an IoT-device, and is used for various purposes, e.g., in industrial surveillance, home surveillance, baby monitors, elderly monitoring, social interaction, movement tracking, etc. This kind of device is often powerful, both in computing and bandwidth, which makes them very attractive for attackers as they can abuse them in additional attacks, such as distributed denial of service (DDoS) attacks. This thesis investigates and presents a few methods used to find and hack IoT-devices. These methods we then apply to IP cameras, where the focus is to examine the impact of these attacks on security and privacy, and to what extent the normal end user can affect (strengthen/weaken) the security. The methods used are based on previously done attacks on IP cameras together with a few other tools used in ethical hacking. The results of the research show that there are vulnerabilities in many of these devices, and that these vulnerabilities have different impacts on security. -

Dissertation Submitted to the Combined Faculties for the Natural
Dissertation submitted to the Combined Faculties for the Natural Sciences and for Mathematics of the Ruperto-Carola University of Heidelberg, Germany for the degree of Doctor of Natural Sciences presented by Sascha Meiers MS B born in Merzig, Germany Date of oral examination: .. Exploiting emerging DNA sequencing technologies to study genomic rearrangements Referees: Dr. Judith Zaugg Prof. Dr. Benedikt Brors Exploiting emerging DNA sequencing technologies to study genomic rearrangements Sascha Meiers th March Supervised by Dr. Jan Korbel Licensed under Creative Commons Attribution (CC BY) . The source code of this thesis is available at https://github.com/meiers/thesis The layout is inspired by and partly taken from Konrad Rudolph’s thesis Summary Structural variants (SVs) alter the structure of chromosomes by deleting, dupli- cating or otherwise rearranging pieces of DNA. They contribute the majority of nucleotide differences between humans and are known to play causal roles in many diseases. Since the advance of massively parallel sequencing (MPS) tech- nologies, SVs have been studied more comprehensively than ever before. How- ever, in contrast to smaller types of genetic variation, SV detection is still funda- mentally hampered by the limitations of short-read sequencing that cannot suf- ficiently cope with the complexity of large genomes. Emerging DNA sequencing technologies and protocols hold the potential to overcome some of these lim- itations. In this dissertation, I present three distinct studies each utilizing such emerging techniques to detect, to validate and/or to characterize SVs. These tech- nologies, together with novel computational approaches that I developed, allow to characterize SVs that had previously been challening, or even impossible, to assess. -

A Tiny Web Server on Embedded System
A Tiny Web Server on Embedded System Lain-Jinn Hwang∗, Tang-Hsun Tu†, Tzu-Hao Lin‡, I-Ting Kuo§, and Bing-Hong Wang¶ Department of Computer Science and Information Engineering Tamkang University, Tamshui, 251, Taipei, Taiwan ∗E-mail: [email protected] †E-mail: war3 [email protected] ‡E-mail: [email protected] §E-mail: [email protected] Abstract [3] has only 64MB RAM and 100MIPS and SiS550 [4] has 128MB RAM and 400MIPS. When we need to build a web Bhttpd (BBS httpd) is a tiny web server for embedded server on these enviroment we have to retrench the size and system, the entire size it consumed is 14KB only. Bhttpd is simplify unnecessary functionality [5]. Usually, the web HTTP/1.1 compliance, GET/POST method, keep-alive con- server built on embedded system is for personal usage. For nection and flow control support. For computer program- example, we can configure the router or switch network set- mer, Bhttpd is easily for reuse because of its structure is ting by browser because there is web server built on it, and simple. In this paper, we introduce the means of bundling we may not often change the setting. Briefly, what we need Bhttpd to make a new web page, by just adding the re- to used on embedded system is a light, small and fit web quired functions. This makes the static web page develop- server. ment on embedded system not only simple but also efficient Usually, web server mean two things, one is a computer than other web server.