Assessing the Diversity and Distribution of Apicomplexans In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Implications of Habitat Restoration for Bumble Bee Population Dynamics, Foraging Ecology, and Epidemiology
IMPLICATIONS OF HABITAT RESTORATION FOR BUMBLE BEE POPULATION DYNAMICS, FORAGING ECOLOGY, AND EPIDEMIOLOGY By Knute Baldwin Gundersen A DISSERTATION Submitted to Michigan State University in partial fulfillment of the requirements for the degree of Entomology—Doctor of Philosophy Ecology, Evolutionary Biology and Behavior—Dual Major 2018 ABSTRACT IMPLICATIONS OF HABITAT RESTORATION FOR BUMBLE BEE POPULATION DYNAMICS, FORAGING ECOLOGY, AND EPIDEMIOLOGY By Knute Baldwin Gundersen Many insects provide valuable ecosystem services, including those that support our food supply. Beneficial insects such as pollinators fulfill part of this role by contributing to approximately one third of the global food crop production. Over the past few decades, pollinators have faced declining populations due to a variety of factors such as agricultural intensification, lack of floral and nesting resources, and disease. One method used in agricultural settings to help sustain pollinator populations is designating unfarmed habitat such as ditches and field margins for habitat enhancement in the form of hedgerows and wildflower strips. These floristically rich areas can be tailored to bloom both before and after crop bloom to help sustain pollinators during the time when crops are not in bloom. In turn, bee populations can benefit from the consistent availability of resources in these areas of habitat enhancement. This dissertation explores how habitat enhancement affects nesting density of a common wild pollinator, Bombus impatiens . Further, this research also aims to determine how foraging preferences change and how bumble bee disease transmission and prevalence respond to habitat enhancement. Research was conducted at 15 commercial highbush blueberry ( Vaccinium corymbosum ) fields in southwest Michigan containing either no restoration, a newly planted restoration, or a mature (5-8 year old) restoration in the field margin from 2015 to 2017. -

New Species of Choleoeimeria (Apicomplexa: Eimeriidae) from The
FOLIA PARASITOLOGICA 53: 91–97, 2006 New species of Choleoeimeria (Apicomplexa: Eimeriidae) from the veiled chameleon, Chamaeleo calyptratus (Sauria: Chamaeleonidae), with taxonomic revision of eimerian coccidia from chameleons Michal Sloboda1 and David Modrý1,2 1Department of Parasitology, University of Veterinary and Pharmaceutical Sciences Brno, Palackého 1–3, 612 42 Brno, Czech Republic; 2Institute of Parasitology, Academy of Sciences of the Czech Republic, Branišovská 31, 370 05 České Budějovice, Czech Republic Key words: Coccidia, Eimeriorina, Choleoeimeria, taxonomy Abstract. Coprological examination of 71 samples from a breeding colony of veiled chameleons, Chamaeleo calyptratus Duméril et Duméril, 1851, revealed a presence of two species of coccidia. In 100% of the samples examined, oocysts of Isospora jaracimrmani Modrý et Koudela, 1995 were detected. A new coccidian species, Choleoeimeria hirbayah sp. n., was discovered in 32.4% of samples from the colony. Its oocysts are tetrasporocystic, cylindrical, 28.3 (25–30) × 14.8 (13.5–17.5) µm, with smooth, bilayered, ~1 µm thick wall. Sporocysts are dizoic, ovoidal to ellipsoidal, 10.1 (9–11) × 6.9 (6–7.5) µm, sporocyst wall is composed of two plates joined by a meridional suture. Endogenous development is confined to the epithelium of the gall blad- der, with infected cells being typically displaced from the epithelium layer towards lumen. A taxonomic revision of tetrasporo- cystic coccidia in the Chamaeleonidae is provided. In reptilian hosts, monoxenous coccidia, namely Ei- of individuals are kept in North America and Europe meria Schneider, 1875 sensu lato and Isospora Schnei- (Nečas 2004). der, 1881 represent commonly diagnosed protozoans of Until now, Isospora jaracimrmani Modrý et Kou- remarkable diversity (Barnard and Upton 1994, Greiner dela, 1995 is the only eimerian coccidium described 2003). -

TEXT-BOOKS of ANIMAL BIOLOGY a General Zoology of The
TEXT-BOOKS OF ANIMAL BIOLOGY * Edited by JULIAN S. HUXLEY, F.R.S. A General Zoology of the Invertebrates by G. S. Carter Vertebrate Zoology by G. R. de Beer Comparative Physiology by L. T. Hogben Animal Ecology by Challes Elton Life in Inland Waters by Kathleen Carpenter The Development of Sex in Vertebrates by F. W. Rogers Brambell * Edited by H. MUNRO Fox, F.R.S. Animal Evolution / by G. S. Carter Zoogeography of the Land and Inland Waters by L. F. de Beaufort Parasitism and Symbiosis by M. Caullery PARASITISM AND ~SYMBIOSIS BY MAURICE CAULLERY Translated by Averil M. Lysaght, M.Sc., Ph.D. SIDGWICK AND JACKSON LIMITED LONDON First Published 1952 !.lADE AND PRINTED IN GREAT BRITAIN BY WILLIAM CLOWES AND SONS, LlMITED, LONDON AND BECCLES CONTENTS LIST OF ILLUSTRATIONS vii PREFACE TO THE ENGLISH EDITION xi CHAPTER I Commensalism Introduction-commensalism in marine animals-fishes and sea anemones-associations on coral reefs-widespread nature of these relationships-hermit crabs and their associates CHAPTER II Commensalism in Terrestrial Animals Commensals of ants and termites-morphological modifications in symphiles-ants.and slavery-myrmecophilous plants . 16 CHAPTER III From Commensalism to Inquilinism and Parasitism Inquilinism-epizoites-intermittent parasites-general nature of modifications produced by parasitism 30 CHAPTER IV Adaptations to Parasitism in Annelids and Molluscs Polychates-molluscs; lamellibranchs; gastropods 40 CHAPTER V Adaptation to Parasitism in the Crustacea Isopoda-families of Epicarida-Rhizocephala-Ascothoracica -

COEVOLUTIONARY RELATIONSHIPS in a NEW TRIPARTITE MARINE SYMBIOSIS Phylogenetic Affinities of the Symbiotic Protist Nephromyces
COEVOLUTIONARY RELATIONSHIPS IN A NEW TRIPARTITE MARINE SYMBIOSIS Phylogenetic affinities of the symbiotic protist Nephromyces Mary Beth Saffo Marine Biological Laboratory, Woods Hole & Museum of Comparative Zoology, Harvard University Abstract 2. Phenotypic features suggestive of general protistan affinities All molgulid ascidian tunicates (Urochordata, phylum Chordata) : •tubular mitochondrial cristae (fig. 5,8) contain, in the lumen of the peculiar, ductless “renal sac,” an equally •thraustochytrid-like sporangia (fig. 4); other developmental stages bear vague peculiar symbiotic protist, Nephromyces. From the first description of resemblances to various other protistan taxa. Nephromyces in the 1870’s, the phylogenetic affinities, even existence, of this taxon has been a matter of debate. Though classified by Alfred Giard in 1888 as a chytridiomycete, Nephromyces has 3. Ssu rDNA sequences indicate that Nephromyces is an apicomplexan clade (preliminary phylogenetic analysis: a sample resisted definitive identification with any particular fungal or protistan Maximum likelihood trees shown) taxa. While its chitinous walls and hyphal-like cells support the notion of fungal affinities for Nephromyces, many other features , including the divergent phylogenetic implications of its morphologically eclectic life cycle , do not. Sequencing of ssu rDNA, indicates, however, that Nephromyces is an apicomplexan; these molecular data are supported by apicomplexan-like characters in the morphology Nephromyces’ host-transfer and infective stages. Other morphological and biochemical features of Nephromyces , some of Phylogenetic analyses, still in progress, consistently indicate thatNephromyces is a distinct them unusual among apicomplexa, or even protists generally, clade within the Apicomplexa. Although the challenge us to consider the significance of certain phenotypic relationship of Nephromyces to other apicomplexan characters as diagnostic tools in inferring deep phylogenies. -

Journal of Parasitology
Journal of Parasitology Eimeria taggarti n. sp., a Novel Coccidian (Apicomplexa: Eimeriorina) in the Prostate of an Antechinus flavipes --Manuscript Draft-- Manuscript Number: 17-111R1 Full Title: Eimeria taggarti n. sp., a Novel Coccidian (Apicomplexa: Eimeriorina) in the Prostate of an Antechinus flavipes Short Title: Eimeria taggarti n. sp. in Prostate of Antechinus flavipes Article Type: Regular Article Corresponding Author: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc University of Melbourne Melbourne, Victoria AUSTRALIA Corresponding Author Secondary Information: Corresponding Author's Institution: University of Melbourne Corresponding Author's Secondary Institution: First Author: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc First Author Secondary Information: Order of Authors: Jemima Amery-Gale, BVSc(Hons), BAnSci, MVSc Joanne Maree Devlin, BVSc(Hons), MVPHMgt, PhD Liliana Tatarczuch David Augustine Taggart David J Schultz Jenny A Charles Ian Beveridge Order of Authors Secondary Information: Abstract: A novel coccidian species was discovered in the prostate of an Antechinus flavipes (yellow-footed antechinus) in South Australia, during the period of post-mating male antechinus immunosuppression and mortality. This novel coccidian is unusual because it develops extra-intestinally and sporulates endogenously within the prostate gland of its mammalian host. Histological examination of prostatic tissue revealed dense aggregations of spherical and thin-walled tetrasporocystic, dizoic sporulated coccidian oocysts within tubular lumina, with unsporulated oocysts and gamogonic stages within the cytoplasm of glandular epithelial cells. This coccidian was observed occurring concurrently with dasyurid herpesvirus 1 infection of the antechinus' prostate. Eimeria- specific 18S small subunit ribosomal DNA PCR amplification was used to obtain a partial 18S rDNA nucleotide sequence from the antechinus coccidian. -

Protocols for Monitoring Harmful Algal Blooms for Sustainable Aquaculture and Coastal Fisheries in Chile (Supplement Data)
Protocols for monitoring Harmful Algal Blooms for sustainable aquaculture and coastal fisheries in Chile (Supplement data) Provided by Kyoko Yarimizu, et al. Table S1. Phytoplankton Naming Dictionary: This dictionary was constructed from the species observed in Chilean coast water in the past combined with the IOC list. Each name was verified with the list provided by IFOP and online dictionaries, AlgaeBase (https://www.algaebase.org/) and WoRMS (http://www.marinespecies.org/). The list is subjected to be updated. Phylum Class Order Family Genus Species Ochrophyta Bacillariophyceae Achnanthales Achnanthaceae Achnanthes Achnanthes longipes Bacillariophyta Coscinodiscophyceae Coscinodiscales Heliopeltaceae Actinoptychus Actinoptychus spp. Dinoflagellata Dinophyceae Gymnodiniales Gymnodiniaceae Akashiwo Akashiwo sanguinea Dinoflagellata Dinophyceae Gymnodiniales Gymnodiniaceae Amphidinium Amphidinium spp. Ochrophyta Bacillariophyceae Naviculales Amphipleuraceae Amphiprora Amphiprora spp. Bacillariophyta Bacillariophyceae Thalassiophysales Catenulaceae Amphora Amphora spp. Cyanobacteria Cyanophyceae Nostocales Aphanizomenonaceae Anabaenopsis Anabaenopsis milleri Cyanobacteria Cyanophyceae Oscillatoriales Coleofasciculaceae Anagnostidinema Anagnostidinema amphibium Anagnostidinema Cyanobacteria Cyanophyceae Oscillatoriales Coleofasciculaceae Anagnostidinema lemmermannii Cyanobacteria Cyanophyceae Oscillatoriales Microcoleaceae Annamia Annamia toxica Cyanobacteria Cyanophyceae Nostocales Aphanizomenonaceae Aphanizomenon Aphanizomenon flos-aquae -

ABSTRACT Gregarine Parasitism in Dragonfly Populations of Central
ABSTRACT Gregarine Parasitism in Dragonfly Populations of Central Texas with an Assessment of Fitness Costs in Erythemis simplicicollis Jason L. Locklin, Ph.D. Mentor: Darrell S. Vodopich, Ph.D. Dragonfly parasites are widespread and frequently include gregarines (Phylum Apicomplexa) in the gut of the host. Gregarines are ubiquitous protozoan parasites that infect arthropods worldwide. More than 1,600 gregarine species have been described, but only a small percentage of invertebrates have been surveyed for these apicomplexan parasites. Some consider gregarines rather harmless, but recent studies suggest otherwise. Odonate-gregarine studies have more commonly involved damselflies, and some have considered gregarines to rarely infect dragonflies. In this study, dragonfly populations were surveyed for gregarines and an assessment of fitness costs was made in a common and widespread host species, Erythemis simplicicollis. Adult dragonfly populations were surveyed weekly at two reservoirs in close proximity to one another and at a flow-through wetland system. Gregarine prevalences and intensities were compared within host populations between genders, among locations, among wing loads, and through time. Host fitness parameters measured included wing load, egg size, clutch size, and total egg count. Of the 37 dragonfly species surveyed, 14 species (38%) hosted gregarines. Thirteen of those species were previously unreported as hosts. Gregarine prevalences ranged from 2% – 52%. Intensities ranged from 1 – 201. Parasites were aggregated among their hosts. Gregarines were found only in individuals exceeding a minimum wing load, indicating that gregarines are likely not transferred from the naiad to adult during emergence. Prevalence and intensity exhibited strong seasonality during both years at one of the reservoirs, but no seasonal trend was detected at the wetland. -

Statistical Comparison of Excystation Methods in Cryptosporidium Parvum Oocysts
MASARYK UNIVERSITY FACULTY OF SCIENCE DEPT . OF BOTANY AND ZOOLOG Y BIOLOGICAL LY ACTIVE COMPOUNDS WIT H POTENTIAL ANTIPARASI TIC EFFECT AND THEIR IMPACT ON THE COURSE OF SELECTED PARASITOSES Ph.D. Dissertation Radka Pecková Supervisor: MVDr. Ivona Foitová, Ph.D . Brno 2018 Bibliographic Entry Author Mgr. Radka Pecková Faculty of Science, Masaryk University Department of Botany and Zoology Biological ly active compounds with potential Title of Thesis: antiparasitic effect and their impact on the processes of selected parasitoses Degree programme: Biology Field of Study: Parasitology Supervisor: MVDr. Ivona Foitová, Ph.D. Academic Year: 2017/2018 Number of Pages: 139 Keywords: Giardia intestinalis ; Cryptosporidium ; Anti - protozoal activity; Plant extracts; Drug of Choice; Natural Antiparasitics; Parasites; Archidendron fagifolium ; Diospyros sumatrana ; Piper betle ; Shorea sumatrana Bibliografický záznam Autor: Mgr. Radka Pecková Přírodovědecká fakulta, Masarykova univerzita Ústav botaniky a zoologie Biologicky aktivní látky s potencionálním Název práce: antiparazitárním účinkem a jejich působení na průběh vybraných parazitóz Studijní program: Biologie Studijní obor: Parazitologie Vedoucí práce: MVDr. Ivona Foitová, Ph.D. Akademický rok: 2017/2018 Počet stran: 139 Klíčová slova: Giardia intestinalis ; Cryptosporidium ; Antiprotozoární aktivita; Rostlinné extrakty; Alternativní léčiva; Přírodní antiparazitika; Paraziti; Archidendron fagifolium ; Diospyros sumatrana ; Piper betle ; Shorea sumatrana ABSTRAK T ABSTRACT This thesis deals with the study of the influence of extracts of selected plants from Indonesia on parasites Giardia intestinalis (Lambl) Alexeieff, 1914 and Cryptosporidium proliferans Kváč, Havrdová, Hlásková, Daňková, Kanděra, Ježková, Vítovec, Sak, Ortega, Xiao, Modrý, Jesudoss Chelladural, Prantlová & McEvoy, 2016 . Tested plants were selected based on behavioural data and the ability to reduce the intensity of parasitic infection in Sumatran orangutans. -
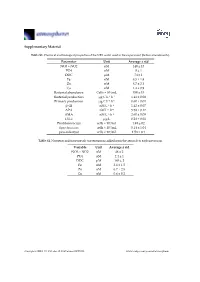
Supplementary Material Parameter Unit Average ± Std NO3 + NO2 Nm
Supplementary Material Table S1. Chemical and biological properties of the NRS water used in the experiment (before amendments). Parameter Unit Average ± std NO3 + NO2 nM 140 ± 13 PO4 nM 8 ± 1 DOC μM 74 ± 1 Fe nM 8.5 ± 1.8 Zn nM 8.7 ± 2.1 Cu nM 1.4 ± 0.9 Bacterial abundance Cells × 104/mL 350 ± 15 Bacterial production μg C L−1 h−1 1.41 ± 0.08 Primary production μg C L−1 h−1 0.60 ± 0.01 β-Gl nM L−1 h−1 1.42 ± 0.07 APA nM L−1 h−1 5.58 ± 0.17 AMA nM L−1·h−1 2.60 ± 0.09 Chl-a μg/L 0.28 ± 0.01 Prochlorococcus cells × 104/mL 1.49 ± 02 Synechococcus cells × 104/mL 5.14 ± 1.04 pico-eukaryot cells × 103/mL 1.58 × 0.1 Table S2. Nutrients and trace metals concentrations added from the aerosols to each mesocosm. Variable Unit Average ± std NO3 + NO2 nM 48 ± 2 PO4 nM 2.4 ± 1 DOC μM 165 ± 2 Fe nM 2.6 ± 1.5 Zn nM 6.7 ± 2.5 Cu nM 0.6 ± 0.2 Atmosphere 2019, 10, 358; doi:10.3390/atmos10070358 www.mdpi.com/journal/atmosphere Atmosphere 2019, 10, 358 2 of 6 Table S3. ANOVA test results between control, ‘UV-treated’ and ‘live-dust’ treatments at 20 h or 44 h, with significantly different values shown in bold. ANOVA df Sum Sq Mean Sq F Value p-value Chl-a 20 H 2, 6 0.03, 0.02 0.02, 0 4.52 0.0634 44 H 2, 6 0.02, 0 0.01, 0 23.13 0.002 Synechococcus Abundance 20 H 2, 7 8.23 × 107, 4.11 × 107 4.11 × 107, 4.51 × 107 0.91 0.4509 44 H 2, 7 5.31 × 108, 6.97 × 107 2.65 × 108, 1.16 × 107 22.84 0.0016 Prochlorococcus Abundance 20 H 2, 8 4.22 × 107, 2.11 × 107 2.11 × 107, 2.71 × 106 7.77 0.0216 44 H 2, 8 9.02 × 107, 1.47 × 107 4.51 × 107, 2.45 × 106 18.38 0.0028 Pico-eukaryote -

Phylogeny and Evolution of Theileria and Babesia Parasites in Selected Wild Herbivores of Kenya
PHYLOGENY AND EVOLUTION OF THEILERIA AND BABESIA PARASITES IN SELECTED WILD HERBIVORES OF KENYA LUCY WAMUYU WAHOME MASTER OF SCIENCE (Bioinformatics and Molecular Biology) JOMO KENYATTA UNIVERSITY OF AGRICULTURE AND TECHNOLOGY 2016 Phylogeny and Evolution of Theileria and Babesia Parasites in Selected Wild Herbivores of Kenya Lucy Wamuyu Wahome A Thesis Submitted in partial fulfillment for the Degree of Master of Science in Bioinformatics and Molecular Biology in the Jomo Kenyatta University of Agriculture and Technology. 2016 DECLARATION This thesis is my original work and has not been presented for a degree in any other University. Signature………………………. Date…………………………… Wahome Lucy Wamuyu This thesis has been submitted for examination with our approval as university supervisors. Signature……………………… Date…………………………… Prof. Daniel Kariuki JKUAT, Kenya Signature……………………… Date…………………………… Dr.Sheila Ommeh JKUAT, Kenya Signature……………………… Date………………………… Dr. Francis Gakuya Kenya Wildlife Service, Kenya ii DEDICATION This work is dedicated to my beloved parents Mr. Godfrey, Mrs. Naomi and my only beloved sister Caroline for their tireless support throughout my academic journey. "I can do everything through Christ who gives me strength." iii ACKNOWLEDGEMENTS Foremost I would like to acknowledge and thank the Almighty God for blessing, protecting and guiding me throughout this period. I am most grateful to the department of Biochemistry, Jomo Kenyatta University of Agriculture and Technology, for according me the opportunity to pursue my postgraduate studies. I would like to express my sincere gratitude to my supervisors Prof. Daniel Kariuki, Dr. Sheila Ommeh and Dr. Francis Gakuya for the useful comments, remarks and engagement through out this project. My deepest gratitude goes to Dr. -

Evidence for and Against Deformed Wing Virus Spillover from Honey Bees to Bumble Bees: a Reverse Genetic Analysis Olesya N
www.nature.com/scientificreports OPEN Evidence for and against deformed wing virus spillover from honey bees to bumble bees: a reverse genetic analysis Olesya N. Gusachenko1*, Luke Woodford1, Katharin Balbirnie‑Cumming1, Eugene V. Ryabov2 & David J. Evans1* Deformed wing virus (DWV) is a persistent pathogen of European honey bees and the major contributor to overwintering colony losses. The prevalence of DWV in honey bees has led to signifcant concerns about spillover of the virus to other pollinating species. Bumble bees are both a major group of wild and commercially‑reared pollinators. Several studies have reported pathogen spillover of DWV from honey bees to bumble bees, but evidence of a sustained viral infection characterized by virus replication and accumulation has yet to be demonstrated. Here we investigate the infectivity and transmission of DWV in bumble bees using the buf-tailed bumble bee Bombus terrestris as a model. We apply a reverse genetics approach combined with controlled laboratory conditions to detect and monitor DWV infection. A novel reverse genetics system for three representative DWV variants, including the two master variants of DWV—type A and B—was used. Our results directly confrm DWV replication in bumble bees but also demonstrate striking resistance to infection by certain transmission routes. Bumble bees may support DWV replication but it is not clear how infection could occur under natural environmental conditions. Deformed wing virus (DWV) is a widely established pathogen of the European honey bee, Apis mellifera. In synergistic action with its vector—the parasitic mite Varroa destructor—it has had a devastating impact on the health of honey bee colonies globally1,2. -

Protistology an International Journal Vol
Protistology An International Journal Vol. 10, Number 2, 2016 ___________________________________________________________________________________ CONTENTS INTERNATIONAL SCIENTIFIC FORUM «PROTIST–2016» Yuri Mazei (Vice-Chairman) Welcome Address 2 Organizing Committee 3 Organizers and Sponsors 4 Abstracts 5 Author Index 94 Forum “PROTIST-2016” June 6–10, 2016 Moscow, Russia Website: http://onlinereg.ru/protist-2016 WELCOME ADDRESS Dear colleagues! Republic) entitled “Diplonemids – new kids on the block”. The third lecture will be given by Alexey The Forum “PROTIST–2016” aims at gathering Smirnov (Saint Petersburg State University, Russia): the researchers in all protistological fields, from “Phylogeny, diversity, and evolution of Amoebozoa: molecular biology to ecology, to stimulate cross- new findings and new problems”. Then Sandra disciplinary interactions and establish long-term Baldauf (Uppsala University, Sweden) will make a international scientific cooperation. The conference plenary presentation “The search for the eukaryote will cover a wide range of fundamental and applied root, now you see it now you don’t”, and the fifth topics in Protistology, with the major focus on plenary lecture “Protist-based methods for assessing evolution and phylogeny, taxonomy, systematics and marine water quality” will be made by Alan Warren DNA barcoding, genomics and molecular biology, (Natural History Museum, United Kingdom). cell biology, organismal biology, parasitology, diversity and biogeography, ecology of soil and There will be two symposia sponsored by ISoP: aquatic protists, bioindicators and palaeoecology. “Integrative co-evolution between mitochondria and their hosts” organized by Sergio A. Muñoz- The Forum is organized jointly by the International Gómez, Claudio H. Slamovits, and Andrew J. Society of Protistologists (ISoP), International Roger, and “Protists of Marine Sediments” orga- Society for Evolutionary Protistology (ISEP), nized by Jun Gong and Virginia Edgcomb.