Studies on Cellular Nutrient Responses and Protein Degradation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phospholipase C-Related Catalytically Inactive Protein: a Novel Signaling Molecule for Modulating Fat Metabolism and Energy Expenditure
Journal of Oral Biosciences 61 (2019) 65e72 Contents lists available at ScienceDirect Journal of Oral Biosciences journal homepage: www.elsevier.com/locate/job Review Phospholipase C-related catalytically inactive protein: A novel signaling molecule for modulating fat metabolism and energy expenditure * Takashi Kanematsu a, b, , Kana Oue a, c, Toshiya Okumura a, Kae Harada a, 1, Yosuke Yamawaki a, 2, Satoshi Asano a, Akiko Mizokami d, Masahiro Irifune c, Masato Hirata e a Department of Cellular and Molecular Pharmacology, Division of Basic Life Sciences, Institute of Biomedical and Health Sciences, Hiroshima University, Hiroshima, 734-8553, Japan b Department of Cell Biology and Pharmacology, Faculty of Dental Science, Kyushu University, Fukuoka, 812-8582, Japan c Department of Dental Anesthesiology, Division of Applied Life Sciences, Institute of Biomedical and Health Sciences, Hiroshima University, Hiroshima, 734- 8553, Japan d OBT Research Center, Faculty of Dental Science, Kyushu University, Fukuoka, 812-8582, Japan e Fukuoka Dental College, Fukuoka, 814-0193, Japan article info abstract Article history: Background: Overweight and obesity are defined as excessive or abnormal fat accumulation in adipose Received 16 March 2019 tissues, and increase the risk of morbidity in many diseases, including hypertension, dyslipidemia, type 2 Received in revised form diabetes, coronary heart disease, and stroke, through pathophysiological mechanisms. There is strong 17 April 2019 evidence that weight loss reduces the risk of metabolic syndrome by limiting blood pressure and Accepted 19 April 2019 improving the levels of serum triglycerides, total cholesterol, low-density lipoprotein-cholesterol, and Available online 15 May 2019 high-density lipoprotein-cholesterol. To date, several attempts have been made to develop effective anti- obesity medication or weight-loss drugs; however, satisfactory drugs for clinical use have not yet been Keywords: Adipose tissue developed. -

The Protein Phosphatase PP2A Plays Multiple Roles in Plant Development by Regulation of Vesicle Traffic—Facts and Questions
International Journal of Molecular Sciences Review The Protein Phosphatase PP2A Plays Multiple Roles in Plant Development by Regulation of Vesicle Traffic—Facts and Questions Csaba Máthé *, Márta M-Hamvas, Csongor Freytag and Tamás Garda Department of Botany, Faculty of Science and Technology, University of Debrecen, H-4032 Debrecen, Hungary; [email protected] (M.M.-H.); [email protected] (C.F.); [email protected] (T.G.) * Correspondence: [email protected] Abstract: The protein phosphatase PP2A is essential for the control of integrated eukaryotic cell functioning. Several cellular and developmental events, e.g., plant growth regulator (PGR) mediated signaling pathways are regulated by reversible phosphorylation of vesicle traffic proteins. Reviewing present knowledge on the relevant role of PP2A is timely. We discuss three aspects: (1) PP2A regulates microtubule-mediated vesicle delivery during cell plate assembly. PP2A dephosphorylates members of the microtubule associated protein family MAP65, promoting their binding to microtubules. Regulation of phosphatase activity leads to changes in microtubule organization, which affects vesicle traffic towards cell plate and vesicle fusion to build the new cell wall between dividing cells. (2) PP2A-mediated inhibition of target of rapamycin complex (TORC) dependent signaling pathways contributes to autophagy and this has possible connections to the brassinosteroid signaling pathway. (3) Transcytosis of vesicles transporting PIN auxin efflux carriers. PP2A regulates vesicle localization and recycling of PINs related to GNOM (a GTP–GDP exchange factor) mediated pathways. The proper intracellular traffic of PINs is essential for auxin distribution in the plant body, thus in whole Citation: Máthé, C.; M-Hamvas, M.; plant development. -

Analyses of PDE-Regulated Phosphoproteomes Reveal Unique and Specific Camp-Signaling Modules in T Cells
Analyses of PDE-regulated phosphoproteomes reveal unique and specific cAMP-signaling modules in T cells Michael-Claude G. Beltejara, Ho-Tak Laua, Martin G. Golkowskia, Shao-En Onga, and Joseph A. Beavoa,1 aDepartment of Pharmacology, University of Washington, Seattle, WA 98195 Contributed by Joseph A. Beavo, May 28, 2017 (sent for review March 10, 2017; reviewed by Paul M. Epstein, Donald H. Maurice, and Kjetil Tasken) Specific functions for different cyclic nucleotide phosphodiester- to bias T-helper polarization toward Th2, Treg, or Th17 pheno- ases (PDEs) have not yet been identified in most cell types. types (13, 14). In a few cases increased cAMP may even potentiate Conventional approaches to study PDE function typically rely on the T-cell activation signal (15), particularly at early stages of measurements of global cAMP, general increases in cAMP- activation. Recent MS-based proteomic studies have been useful dependent protein kinase (PKA), or the activity of exchange in characterizing changes in the phosphoproteome of T cells under protein activated by cAMP (EPAC). Although newer approaches various stimuli such as T-cell receptor stimulation (16), prosta- using subcellularly targeted FRET reporter sensors have helped glandin signaling (17), and oxidative stress (18), so much of the define more compartmentalized regulation of cAMP, PKA, and total Jurkat phosphoproteome is known. Until now, however, no EPAC, they have limited ability to link this regulation to down- information on the regulation of phosphopeptides by PDEs has stream effector molecules and biological functions. To address this been available in these cells. problem, we have begun to use an unbiased mass spectrometry- Inhibitors of cAMP PDEs are useful tools to study PKA/EPAC- based approach coupled with treatment using PDE isozyme- mediated signaling, and selective inhibitors for each of the 11 PDE – selective inhibitors to characterize the phosphoproteomes of the families have been developed (19 21). -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

Protein Interactomes of Protein Phosphatase 2A B55 Regulatory
www.nature.com/scientificreports OPEN Protein interactomes of protein phosphatase 2A B55 regulatory subunits reveal B55-mediated Received: 22 September 2017 Accepted: 24 January 2018 regulation of replication protein A Published: xx xx xxxx under replication stress Feifei Wang1,2, Songli Zhu2, Laura A. Fisher2, Weidong Wang1, Gregory G. Oakley2, Chunling Li1 & Aimin Peng2 The specifc function of PP2A, a major serine/threonine phosphatase, is mediated by regulatory targeting subunits, such as members of the B55 family. Although implicated in cell division and other pathways, the specifc substrates and functions of B55 targeting subunits are largely undefned. In this study we identifed over 100 binding proteins of B55α and B55β in Xenopus egg extracts that are involved in metabolism, mitochondria function, molecular trafcking, cell division, cytoskeleton, DNA replication, DNA repair, and cell signaling. Among the B55α and B55β-associated proteins were numerous mitotic regulators, including many substrates of CDK1. Consistently, upregulation of B55α accelerated M-phase exit and inhibited M-phase entry. Moreover, specifc substrates of CDK2, including factors of DNA replication and chromatin remodeling were identifed within the interactomes of B55α and B55β, suggesting a role for these phosphatase subunits in DNA replication. In particular, we confrmed in human cells that B55α binds RPA and mediates the dephosphorylation of RPA2. The B55-RPA association is disrupted after replication stress, consistent with the induction of RPA2 phosphorylation. Thus, we report here a new mechanism that accounts for both how RPA phosphorylation is modulated by PP2A and how the phosphorylation of RPA2 is abruptly induced after replication stress. Protein phosphorylation, a major form of post-translational modifcation, plays a crucial role in regulation of pro- tein functions. -

Protein Phosphatases in N Response and NUE in Crops
Chapter 12 Protein Phosphatases in N Response and NUE in Crops Supriya Kumari and Nandula Raghuram 12.1 Introduction Nitrogen is an essential macronutrient for plant growth and crop yield, as most crop plants barring N-fixing legumes depend on reactive N compounds such as urea, ammonium salts, or nitrates from manures or fertilizers as their main nitrogen source. Fertilizers are the world’s largest anthropogenic source of reactive nitrogen, with annual global consumption at approximately 119.40 million tons and annual growth of 1.4% (FAO 2018). Due to the poor N use efficiency (NUE) of most crops at a global average of <30%, unused reactive N compounds from fertilizers and other sources are a major source of pollution of land, water, and air, affecting human and animal health, biodiversity, and climate change (Sutton and Bleeker 2013; Sutton et al. 2019).The fourth UN Environment Assembly held in March 2019 has adopted a resolution to work toward sustainable nitrogen management (https://sdg. iisd.org/events/fourth-session-of-the-un-environment-assembly-unea-4/). Therefore, the development of N use efficient crops is an extremely important goal, not only for sustainable agriculture but also for environmental sustainability and mitigation of climate change (Raghuram et al. 2007; Raghuram and Sharma 2019). NUE can be defined in several ways (Raghuram and Sharma 2019), but it is best understood as yield per unit N, or the maximal output with minimal N input. NUE is a complex genetic trait involving several genes and their interactions with the environment, which are yet to be fully characterized (Mandal et al. -
![RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)](https://docslib.b-cdn.net/cover/9005/rt%C2%B2-profiler-pcr-array-96-well-format-and-384-well-4-x-96-format-1459005.webp)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 x 96] Format) Human Protein Phosphatases Cat. no. 330231 PAHS-045ZA For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Applied Biosystems® models 5700, 7000, 7300, 7500, Format A 7700, 7900HT, ViiA™ 7 (96-well block); Bio-Rad® models iCycler®, iQ™5, MyiQ™, MyiQ2; Bio-Rad/MJ Research Chromo4™; Eppendorf® Mastercycler® ep realplex models 2, 2s, 4, 4s; Stratagene® models Mx3005P®, Mx3000P®; Takara TP-800 RT² Profiler PCR Array, Applied Biosystems models 7500 (Fast block), 7900HT (Fast Format C block), StepOnePlus™, ViiA 7 (Fast block) RT² Profiler PCR Array, Bio-Rad CFX96™; Bio-Rad/MJ Research models DNA Format D Engine Opticon®, DNA Engine Opticon 2; Stratagene Mx4000® RT² Profiler PCR Array, Applied Biosystems models 7900HT (384-well block), ViiA 7 Format E (384-well block); Bio-Rad CFX384™ RT² Profiler PCR Array, Roche® LightCycler® 480 (96-well block) Format F RT² Profiler PCR Array, Roche LightCycler 480 (384-well block) Format G RT² Profiler PCR Array, Fluidigm® BioMark™ Format H Sample & Assay Technologies Description The Human Protein Phosphatases RT² Profiler PCR Array profiles the gene expression of the 84 most important and well-studied phosphatases in the mammalian genome. By reversing the phosphorylation of key regulatory proteins mediated by protein kinases, phosphatases serve as a very important complement to kinases and attenuate activated signal transduction pathways. The gene classes on this array include both receptor and non-receptor tyrosine phosphatases, catalytic subunits of the three major protein phosphatase gene families, the dual specificity phosphatases, as well as cell cycle regulatory and other protein phosphatases. -
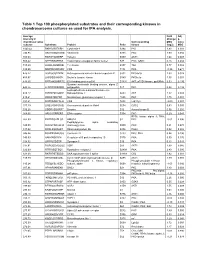
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Analyses of PDE-Regulated Phosphoproteomes Reveal Unique and Specific Camp Signaling Modules in T Cells
Analyses of PDE-regulated phosphoproteomes reveal unique and specific cAMP signaling modules in T cells Michael-Claude G. Beltejar A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2017 Reading Committee: Joseph Beavo, Chair Shao-en Ong Richard Gardner Program Authorized to Offer Degree: Molecular and Cellular Biology ©Copyright 2017 Michael-Claude G. Beltejar University of Washington Abstract Analyses of PDE-regulated phosphoproteomes reveal unique and specific cAMP signaling modules in T cells Michael-Claude G. Beltejar Chair of the Supervisory Committee: Professor Joseph A. Beavo Department of Pharmacology Specific functions for different cyclic nucleotide phosphodiesterases (PDEs) have not yet been identified in most cell types. Conventional approaches to study PDE function typically rely on measurements of global cAMP, general increases in cAMP-dependent protein kinase (PKA), or exchange protein activated by cAMP (EPAC) activity. Although newer approaches utilizing subcellularly-targeted FRET reporter sensors have helped to define more compartmentalized regulation of cAMP, PKA, and EPAC, they have limited ability to link this regulation to downstream effector molecules and biological functions. To address this problem, we used an unbiased, mass spectrometry based approach coupled with treatment using PDE isozyme- selective inhibitors to characterize the phosphoproteomes of the “functional pools” of cAMP/PKA/EPAC that are regulated by specific cAMP-PDEs (the PDE-regulated phosphoproteomes). In Jurkat cells we found multiple, distinct phosphoproteomes that differ in response to different PDE inhibitors. We also found that little phosphorylation occurs unless at least 2 different PDEs are concurrently inhibited in these cells. Moreover, bioinformatics analyses of these phosphoproteomes provides insight into the functional roles, mechanisms of action, and synergistic relationships among the different PDEs that coordinate cAMP-signaling cascades in these cells. -

Therapeutic Opportunities in Colon Cancer Focus on Phosphodiesterase Inhibitors
Life Sciences 230 (2019) 150–161 Contents lists available at ScienceDirect Life Sciences journal homepage: www.elsevier.com/locate/lifescie Review article Therapeutic opportunities in colon cancer: Focus on phosphodiesterase inhibitors T ⁎ Ankita Mehta, Bhoomika M. Patel Institute of Pharmacy, Nirma University, Ahmedabad, India ARTICLE INFO ABSTRACT Keywords: Despite novel technologies, colon cancer remains undiagnosed and 25% of patients are diagnosed with meta- Phosphodiesterases static colon cancer. Resistant to chemotherapeutic agents is one of the major problems associated with treating cAMP colon cancer which creates the need to develop novel agents targeting towards newer targets. A phosphodies- cGMP terase is a group of isoenzyme, which, hydrolyze cyclic nucleotides and thereby lowers intracellular levels of Adenylate cyclase cAMP and cGMP leading to tumorigenic effects. Many in vitro and in vivo studies have confirmed increased PDE Guanylate cyclase expression in different types of cancers including colon cancer. cAMP-specific PDE inhibitors increase in- Colon cancer tracellular cAMP that leads to activation of effector molecules-cAMP-dependent protein kinase A, exchange protein activated by cAMP and cAMP gated ion channels. These molecules regulate cellular responses and exert its anticancer role through different mechanisms including apoptosis, inhibition of angiogenesis, upregulating tumor suppressor genes and suppressing oncogenes. On the other hand, cGMP specific PDE inhibitors exhibit anticancer effects through cGMP dependent protein kinase and cGMP dependent cation channels. Elevation in cGMP works through activation of caspases, suppression of Wnt/b-catenin pathway and TCF transcription leading to inhibition of CDK and survivin. These studies point out towards the fact that PDE inhibition is as- sociated with anti-proliferative, anti-apoptotic and anti-angiogenic pathways involved in its anticancer effects in colon cancer. -

(12) Patent Application Publication (10) Pub. No.: US 2003/0198970 A1 Roberts (43) Pub
US 2003O19897OA1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0198970 A1 Roberts (43) Pub. Date: Oct. 23, 2003 (54) GENOSTICS clinical trials on groups or cohorts of patients. This group data is used to derive a Standardised method of treatment (75) Inventor: Gareth Wyn Roberts, Cambs (GB) which is Subsequently applied on an individual basis. There is considerable evidence that a significant factor underlying Correspondence Address: the individual variability in response to disease, therapy and FINNEGAN, HENDERSON, FARABOW, prognosis lies in a person's genetic make-up. There have GARRETT & DUNNER been numerous examples relating that polymorphisms LLP within a given gene can alter the functionality of the protein 1300 ISTREET, NW encoded by that gene thus leading to a variable physiological WASHINGTON, DC 20005 (US) response. In order to bring about the integration of genomics into medical practice and enable design and building of a (73) Assignee: GENOSTIC PHARMA LIMITED technology platform which will enable the everyday practice (21) Appl. No.: 10/206,568 of molecular medicine a way must be invented for the DNA Sequence data to be aligned with the identification of genes (22) Filed: Jul. 29, 2002 central to the induction, development, progression and out come of disease or physiological States of interest. Accord Related U.S. Application Data ing to the invention, the number of genes and their configu rations (mutations and polymorphisms) needed to be (63) Continuation of application No. 09/325,123, filed on identified in order to provide critical clinical information Jun. 3, 1999, now abandoned. concerning individual prognosis is considerably less than the 100,000 thought to comprise the human genome. -

Live-Cell Imaging Rnai Screen Identifies PP2A–B55α and Importin-Β1 As Key Mitotic Exit Regulators in Human Cells
LETTERS Live-cell imaging RNAi screen identifies PP2A–B55α and importin-β1 as key mitotic exit regulators in human cells Michael H. A. Schmitz1,2,3, Michael Held1,2, Veerle Janssens4, James R. A. Hutchins5, Otto Hudecz6, Elitsa Ivanova4, Jozef Goris4, Laura Trinkle-Mulcahy7, Angus I. Lamond8, Ina Poser9, Anthony A. Hyman9, Karl Mechtler5,6, Jan-Michael Peters5 and Daniel W. Gerlich1,2,10 When vertebrate cells exit mitosis various cellular structures can contribute to Cdk1 substrate dephosphorylation during vertebrate are re-organized to build functional interphase cells1. This mitotic exit, whereas Ca2+-triggered mitotic exit in cytostatic-factor- depends on Cdk1 (cyclin dependent kinase 1) inactivation arrested egg extracts depends on calcineurin12,13. Early genetic studies in and subsequent dephosphorylation of its substrates2–4. Drosophila melanogaster 14,15 and Aspergillus nidulans16 reported defects Members of the protein phosphatase 1 and 2A (PP1 and in late mitosis of PP1 and PP2A mutants. However, the assays used in PP2A) families can dephosphorylate Cdk1 substrates in these studies were not specific for mitotic exit because they scored pro- biochemical extracts during mitotic exit5,6, but how this relates metaphase arrest or anaphase chromosome bridges, which can result to postmitotic reassembly of interphase structures in intact from defects in early mitosis. cells is not known. Here, we use a live-cell imaging assay and Intracellular targeting of Ser/Thr phosphatase complexes to specific RNAi knockdown to screen a genome-wide library of protein substrates is mediated by a diverse range of regulatory and targeting phosphatases for mitotic exit functions in human cells. We subunits that associate with a small group of catalytic subunits3,4,17.