Dwarfism Mental Retardation Syndrome (ACD Syndrome): Description of an Additional Case and Review of the Literature
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bilateral Lower Extremity Hyperkeratotic Plaques: a Case Report of Ichthyosis Vulgaris
Faculty & Staff Scholarship 2015 Bilateral lower extremity hyperkeratotic plaques: a case report of ichthyosis vulgaris Hayley Leight Zachary Zinn Omid Jalali Follow this and additional works at: https://researchrepository.wvu.edu/faculty_publications Clinical, Cosmetic and Investigational Dermatology Dovepress open access to scientific and medical research Open Access Full Text Article CASE REPORT Bilateral lower extremity hyperkeratotic plaques: a case report of ichthyosis vulgaris Hayley Leight Abstract: Here, we report a case of a middle-aged woman presenting with severe, long-standing, Zachary Zinn hyperkeratotic plaques of the lower extremities unrelieved by over-the-counter medications. Omid Jalali Initial history and clinical findings were suggestive of an inherited ichthyosis. Ichthyoses are genetic disorders characterized by dry scaly skin and altered skin-barrier function. A diagnosis Department of Dermatology, West Virginia University, of ichthyosis vulgaris was confirmed by histopathology. Etiology, prevalence, and treatment Morgantown, WV, USA options are discussed. Keywords: filaggrin gene, FLG, profilaggrin, keratohyalin granules, hyperkeratosis Introduction For personal use only. Inherited ichthyoses are a diverse group of genetic disorders characterized by dry, scaly skin; hyperkeratosis; and altered skin-barrier function. While these disorders of cutaneous keratinization are multifaceted and varying in etiology, disruption in the stratum corneum with generalized scaling is common to all.1–4 Although not entirely known -

Proceedings of the 16Th Annual Meeting of the Society for Pediatric Dermatoiogy
SPECIAL ARTICLE Pediatric Dermatology Vol. 9 No. 1 66-76 Proceedings of the 16th Annual Meeting of the Society for Pediatric Dermatoiogy WiUiamsburg, Virginia June 3a-July 3, 1991 Eleanor £. Sahn, M.D. Medical University of South Carolina Charleston, South Carolina A. Howiand Hartley, M.D. Children's Hospital National Medical Center Washington, D.C. Stephen Gellis, M.D. Children's Hospital Medical Center Boston, Massachusetts James E. Rasmussen, M.D. University of Michigan Medical Center Ann Arbor, Michigan Monday, July 1, 1991 ture by the newspaper account he received, dated December 17, 1799, telling of General George Dr. Alfred T. Lane (Stanford University) orga- Washington's death. We learn the story of General nized the sixteenth annual meeting of the Society Washington's rapid demise, probably from bacterial for Pediatric Dermatology, held in Wiliiamsburg, infection, hastened by the medical treatments of the Virgitiia. The seventh annual Sidney Hurwitz Lec- day, including frequent and copious blood letting. ture was delivered by Dr. Rona M. MacKie (Uni- There was a current saying, "more people died an- versity of Glasgow) on "Melanoma: Risk Factors in nually from lancets than from swords." Dysplastic Nevus Syndrome." President Anne Lucky (Cincinnati, Ohio) welcomed the society MELANOMA: RISK FACTORS AND members to Wiliiamsburg and introduced the first DYSPLASTIC NEVUS SYNDROME speaker. Dr. Rona MacKie first discussed risk factors in mel- anoma, citing several large case control studies car- COLONIAL MEDICINE ried out in western Canada, Scotland, Scandinavia, Dr. Tor A. Shwayder (Henry Ford Hospital) pre- and Germany. The frequency of melanoma has dou- sented a delightful and professional "Character In- bled each decade in Scandinavia, the United King- terpreter Portrayal of Iseiac Shwayder, Medical dom, and Germany. -

Mutation and Expression of Abca12in Keratosis Pilaris and Nevus
MOLECULAR MEDICINE REPORTS 18: 3153-3158, 2018 Mutation and expression of ABCA12 in keratosis pilaris and nevus comedonicus FEN LIU1,2*, YAO YANG1,3*, YAN ZHENG1,3, YAN-HUA LIANG1,3 and KANG ZENG1 1Department of Dermatology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong 510515; 2Department of Histology and Embryology, Institute of Neuroscience, School of Basic Medical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang 325035; 3Department of Dermatology, Shenzhen Hospital, Southern Medical University, Shenzhen, Guangdong 518100, P.R. China Received June 22, 2017; Accepted April 17, 2018 DOI: 10.3892/mmr.2018.9342 Abstract. Keratosis pilaris (KP) and nevus comedonicus Introduction (NC) are congenital keratinized dermatoses; however, the exact etiology of these two diseases is unclear. The objective Keratosis pilaris (KP; OMIM #604093), also known as of the present study was to identify the disease-causing genes lichen pilaris, is a benign genodermatosis that is estimated and their association with functional alterations in the devel- to effect ~40% of the population (1). KP is characterized by opment of KP and NC. Peripheral blood samples of one KP the presence of symmetric, asymptomatic and grouped kera- family, two NC families and 100 unrelated healthy controls totic follicular papules with varying degrees of perifollicular were collected. The genomic sequences of 147 genes associ- erythema. KP lesions often involve the proximal and extended ated with 143 genetic skin diseases were initially analyzed parts of extremities, the cheeks and the buttocks (2). Cases from the KP proband using a custom-designed GeneChip. A may be generalized or unilateral (2). Most patients develop KP novel heterozygous missense mutation in the ATP-binding in their childhood, with a peak in incidence during adoles- cassette sub-family A member 12 (ABCA12) gene, designated cence (3). -

Genodermatoses
GENODERMATOSES Genodermatoses What’s new? Nigel P Burrows C Filaggrin mutations underlie ichthyosis vulgaris and are a risk factor for atopy including eczema, allergic sensitization, asthma, allergic rhinitis and peanut allergy Abstract C A new classification and nomenclature for ichthyoses was Genetic skin diseases encompass a spectrum from the common to the published in 2009, peeling skin syndromes which may be rare. It is important for the clinician to be alert to the possibility that confused for the milder subtypes of epidermolysis bullosa are the patient may be presenting for the first time with one or more features distinct genetic entities of a genetic disease so that appropriate investigation and counselling can C Emerging evidence that pseudoxanthoma elasticum is a meta- take place. Recent discoveries have helped the understanding of many of bolic disorder resulting in calcification of elastic fibres these disorders. A few common and important genodermatoses are high- C Mammalian target of rapamycin (mTOR) inhibitor therapies are lighted in this article. showing promise in tuberous sclerosis complex C Vascular anomalies on the skin may be the presenting feature of Keywords cancer syndromes; collagen; epidermolysis bullosa; filaggrin; inherited syndromes genodermatoses; ichthyosis; keratinization; pseudoxanthoma elasticum; vascular anomalies X-linked recessive ichthyosis In 75% of cases of X-linked recessive ichthyosis (XLRI) (Figure 1), scaling is present in the first week of life and tends to progress into adolescence. In contrast to IV, the flexures may be Genetic skin diseases encompass a spectrum from the common involved. A third of cases are associated with a prolonged labour. (e.g. atopic eczema) to the rare (e.g. -

Ichthyosis Vulgaris a Case Report and Review of Literature Sarah E
CASE REPORT Ichthyosis Vulgaris A Case Report and Review of Literature Sarah E. Mertz, Thea D. Nguyen, Lori A. Spies chthyosis vulgaris (IV) is a hereditary skin condi- removal surgery. By patient report, the lesions had been tion characterized by an accumulation of cells in present since childhood but progressively worsened over the horny layer that manifests as xerotic, plate- the past few years because of a lack of skin care routine. As like scales. It is most prominent on the extensor is typical in IV, there was a history of improvement of surfaces of the extremities, back, abdomen, and symptoms in warmer months of the summer. Comorbidities legs and exhibits palmar hyperlinearity (Takeichi & to the long-standing IV and cognitive disability include IAkiyama, 2016). If not properly treated, this build up hypertension and obesity. There was no personal history of can cause difficulty in patient care and a decrease in the skin cancer, and the patient was not able to provide details of quality of life of those afflicted with this condition. his family history. On physical examination, significant and There are more than 20 types of ichthyosis to include pertinent findings include diffuse symmetric, thick, hyper- epidermolytic ichthyosis, congential reticular ichthyosiform keratotic, light-gray, fish-scaled plaques with fissures prom- erythroderma, and lamellar ichthyosis, with IV being the inent on the dorsal and ventral surfaces of the bilateral upper most common type of hereditary nonsyndromic ichthyosis extremities including arms, forearms, dorsal hands, and and characterized as a reduction of keratohyalin granules posterior neck and fine, scaly, erythematous areas of or a granular layer absence (Takeichi & Akiyama, 2016). -

Hereditary Ichthyosis
!" #$%&'# $(%&) #'# %*+&,*'#'* -#.*&%* --#.# // Dissertation for Degree of Doctor of Philosophy (Faculty of Medicine) in Dermatology and Venereology presented at Uppsala University in 2002 ABSTRACT Gånemo, A. 2002. Hereditary ichthyosis. Causes, Skin Manifestations, Treatments and Quality of Life. Acta Universitatis Upsaliensis. Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1125. 68 pp Uppsala ISBN 91-554-5246-9 Hereditary ichthyosis is a collective name for many dry and scaly skin disorders ranging in frequency from common to very rare. The main groups are autosomal recessive lamellar ichthyosis, autosomal dominant epidermolytic hyperkeratosis and ichthyosis vulgaris, and x-linked recessive ichthyosis. Anhidrosis, ectropion and keratodermia are common symptoms, especially in lamellar ichthyosis, which is often caused by mutations in the transglutaminase 1 (TGM1) gene. The aim of this work was to study patients with different types of ichthyosis regarding (i) the patho-aetiology (TGM1 and electron microscopy [EM] analysis), (ii) skin signs and symptoms (clinical score and subjective measure of disease activity), (iii) quality of life (questionnaires DLQI, SF-36 and NHP and face-to-face interviews) and (iv) a search for new ways of topical treatment. Patients from Sweden and Estonia with autosomal recessive congenital ichthyosis (n=83) had a broader clinical spectrum than anticipated, but a majority carried TGM1 mutations. Based on DNA analysis and clinical examinations the patients were classified into three groups, which could be further subdivided after EM analysis. Our studies indicate that patients with ichthyosis have reduced quality of life as reflected by DLQI and by some domains of SF- 36, by NHP and the interviews. All the interviewees reported that their skin disease had affected them negatively to varying degrees during their entire lives and that the most problematic period was childhood. -

Keratosis Pilaris: a Common Follicular Hyperkeratosis
PEDIATRIC DERMATOLOGY Series Editor: Camila K. Janniger, MD Keratosis Pilaris: A Common Follicular Hyperkeratosis Sharon Hwang, MD; Robert A. Schwartz, MD, MPH Keratosis pilaris (KP) is a common inherited dis- 155 otherwise unaffected patients.2 In the adolescent order of follicular hyperkeratosis. It is character- population, its prevalence is postulated to be at least ized by small, folliculocentric keratotic papules 50%; it is more common in adolescent females than that may have surrounding erythema. The small males, seen in up to 80% of adolescent females.3 papules impart a stippled appearance to the skin The disorder is inherited in an autosomal domi- resembling gooseflesh. The disorder most com- nant fashion with variable penetrance; no specific monly affects the extensor aspects of the upper gene has been identified. In a study of 49 evaluated arms, upper legs, and buttocks. Patients with KP patients, there was a positive family history of KP in usually are asymptomatic, with complaints limited 19 patients (39%), while 27 patients (55%) had no to cosmetic appearance or mild pruritus. When family history of the disorder.4 diagnosing KP, the clinician should be aware that a number of diseases are associated with KP such Clinical Features as keratosis pilaris atrophicans, erythromelanosis The keratotic follicular papules of KP most commonly follicularis faciei et colli, and ichthyosis vulgaris. are grouped on the extensor aspects of the upper arms Treatment options vary, focusing on avoiding skin (Figure), upper legs, and buttocks.4 Other affected dryness, using emollients, and adding keratolytic locations may include the face and the trunk.5 The agents or topical steroids when necessary. -

The Clinical Spectrum of Congenital
Acta Derm Venereol 2003, Suppl. 213: 34–47 The Clinical Spectrum of Congenital Ichthyosis in Sweden: A Review of 127 Cases ANDERS VAHLQUIST1, AGNETA GÅNEMO1, MARITTA PIGG2, MARIE VIRTANEN1 AND PER WESTERMARK3 Departments of 1Dermatology, 2Clinical Genetics and 3Pathology, University Hospital, Uppsala, Sweden Congenital ichthyosis comprises a rare group of usual- microscopy, HI: Harlequin ichthyosis, IFAP: ichthyosis follicularis, ly monogenetic diseases that present at birth as a collo- alopecia and photofobia, LI-TGM: lamellar ichthyosis with dion phenotype or as variable degrees of ichthyosiform transglutaminase 1 gene mutations, LI/CIE: lamellar ichthyosis and/ or congenital ichthyosiform erythroderma, KID: keratitis, ichthyo- erythroderma, with or without superficial blisters. De- sis and deafness, K: keratin, KLICK: keratosis linearis, ichthyosis pending on which gene mutation causes the disease, the congenita and keratoderma, SLS: Sjögren-Larsson syndrome, Tgase skin problems later in life may range from a severe 1 – transglutaminase 1 protein, XRI: X-linked recessive ichthyosis. lamellar or bullous ichthyosis to mild or only focally expressed hyperkeratotic lesions. It is obviously impor- tant, but sometimes painstakingly difficult, to make a INTRODUCTION correct diagnosis already in infancy. Fortunately, re- cent advances in our understanding of the molecular Congenital ichthyosis (CI) encompasses a large group of genetics of ichthyosis have led to several new diagnos- mostly monogenetic disorders of keratinization present- tic tools that are continuously being updated. Based on ing at birth as widespread hyperkeratosis and scaling of the integument, and sometimes associated with erythro- Downloaded By: [Akademiska Sjukhuset] At: 12:48 5 October 2007 this development, and on our own 5 years of experi- ence in a national genodermatosis centre, we describe derma and skin erosions. -

Genetic Exploration of the Ichthyoses
A DERMATOLOGY FOUNDATION PUBLICATION SPONSORED BY ORTHO DERMATOLOGICS A Division of Valeant Pharmaceuticals North America LLC VOL.DERMATOLOGYDERMATOLOGY 36 NO. 3 FALL 2017 ™ FOCUSFOCUS Also In This Issue DF Welcomes New Annenberg Circle and AC Sustaining Members Genetic Exploration Dr. John E. Harris Leads Medical & Scientific Committee of the Ichthyoses: JAAD Highlights Research Reaping a Multitude of Benefits From DF-Supported Investigators odern next-generation sequencing (NGS) keratinization (DOK), which encompass at M technologies—also known as high- least 28 ichthyoses and other scaly skin disor- Focus on Research throughput DNA sequencing—burst onto the ders that disrupt the skin barrier and thus sig- CAAR-T Cells— scene roughly a decade ago, powering a nificantly impair cutaneous water retention A Revolution in Therapy for revolution in genetics research. It completely (see boxes on pages 8 and 10). These diseases outstripped Moore’s law—normally the have been a career-long passion in both Autoimmune Disease Begins benchmark for measuring technologic the clinic and lab for Keith A. Choate, MD, With Pemphigus Vulgaris advance—which observes that computing PhD, professor of dermatology, genetics, capacity doubles nearly every two years, with and pathology at Yale and director of research Aimee S. Payne, MD, PhD dramatic progress enabling the transforma- in dermatology there. When this game- Albert M. Kligman Associate Professor of Dermatology, Department of Dermatology, Perelman School of tional power of iPhones, personal computers, changing revolution in molecular diagnostics Medicine, University of Pennsylvania and social media. Although advances in DNA emer ged, he was ready to dive in—and has sequencing technology were in line with been a prime mover in this progress from the r. -

Congenital: Growths-Conditions-Vascular Lesions-Hair-Nails Epidermolysis-Ichthyosis-Mastocytosis-Neurofibromatosis-Tuberous Sclerosis-Xeroderma
Congenital: Growths-Conditions-Vascular Lesions-Hair-Nails Epidermolysis-Ichthyosis-Mastocytosis-Neurofibromatosis-Tuberous sclerosis-Xeroderma Other congenital malformations (Q80-Q85) Q80 Congenital ichthyosis Excludes1: Refsum's disease (G60.1) Q80.0 Ichthyosis vulgaris Q80.1 X-linked ichthyosis Q80.2 Lamellar ichthyosis Collodion baby Q80.3 Congenital bullous ichthyosiform erythroderma Q80.4 Harlequin fetus Q80.8 Other congenital ichthyosis Q80.9 Congenital ichthyosis, unspecified Q81 Epidermolysis bullosa Q81.0 Epidermolysis bullosa simplex Excludes1: Cockayne's syndrome (Q87.1) Q81.1 Epidermolysis bullosa letalis Herlitz' syndrome Q81.2 Epidermolysis bullosa dystrophica Q81.8 Other epidermolysis bullosa Q81.9 Epidermolysis bullosa, unspecified Q82 Other congenital malformations of skin Excludes1: acrodermatitis enteropathica (E83.2) congenital erythropoietic porphyria (E80.0) pilonidal cyst or sinus (L05.-) Sturge-Weber (-Dimitri) syndrome (Q85.8) Q82.0 Hereditary lymphedema Q82.1 Xeroderma pigmentosum Q82.2 Mastocytosis Urticaria pigmentosa Excludes1: malignant mastocytosis (C96.2) Q82.3 Incontinentia pigmenti Q82.4 Ectodermal dysplasia (anhidrotic) Excludes1: Ellis-van Creveld syndrome (Q77.6) Q82.5 Congenital non-neoplastic nevus Birthmark NOS Flammeus Nevus Portwine Nevus Sanguineous Nevus Strawberry Angioma Strawberry Nevus Vascular Nevus NOS Verrucous Nevus Excludes2: Café au lait spots (L81.3) lentigo (L81.4) nevus NOS (D22.-) araneus nevus (I78.1) melanocytic nevus (D22.-) pigmented nevus (D22.-) spider nevus (I78.1) stellar -
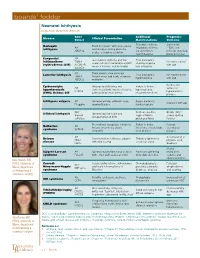
Boards' Fodder
boards’ fodder Neonatal Ichthyosis by Alyx Rosen, MD and Kate Oberlin, MD Gene Additional Prognosis/ Disease Clinical Presentation Defect Manifestations Outcome Premature delivery; Survival rate Harlequin Thick membrane with deep cracks AR respiratory distress; ~50%; oral ichthyosis and fissures forming plate-like ABCA12 ear deformities; retinoids may help scales, ectropion, eclabium fluid imbalance prolong survival Congenital AR Generalized erythema and fine Heat intolerance; ichthyosiform TGM-1 No improvement scale; collodion membrane at birth; scarring alopecia; erythroderma (CIE) ALOX12B with age involves flexures; nail dystrophy rare ectropion ALOXE3 AR Thick, brown scale involving Lamellar ichthyosis Heat intolerance; No improvement TGM-1 flexural areas and trunk; eclabium, hypernatremia with age ABCA12 ectropion Evolves into Epidermolytic Widespread blistering and Failure to thrive; AD verrucous hyperkeratosis erythema at birth; involves flexures, hypernatremia; K1/K10 hyperkeratotic (EHK); Bullous CIE palmoplantar keratoderma recurrent infections plaques Ichthyosis vulgaris AD Generalized fine, adherent scale Atopic diathesis; Improves with age Filaggrin sparing flexures keratosis pilaris XLR Corneal opacities; Brown “dirty” X-linked ichthyosis Generalized fine scale and Steroid cryptorchidism; scales sparing desquamation at birth sulfatase prolonged labor flexures Trichorrhexis invaginata, ichthyosis Failure to thrive; Pruritus; Netherton AR linearis circumflexa, atopic infections; elevated IgE; eczematous syndrome SPINK5 dermatitis -
A Clinical Study on Genodermatoses and Their Effect On
A CLINICAL STUDY ON GENODERMATOSES AND THEIR EFFECT ON QUALITY OF LIFE – PROSPECTIVE OBSERVATIONAL STUDY IN A TERTIARY CARE HOSPITAL Dissertation Submitted to THE TAMILNADU DR.M.G.R. MEDICAL UNIVERSITY IN PARTIAL FULFILMENT FOR THE AWARD OF THE DEGREE OF DOCTOR OF MEDICINE IN DERMATOLOGY, VENEREOLOGY & LEPROSY Register No.: 201730253 BRANCH XX MAY 2020 DEPARTMENT OF DERMATOLOGY VENEREOLOGY & LEPROSY TIRUNELVELI MEDICAL COLLEGE TIRUNELVELI -11 BONAFIDE CERTIFICATE This is to certify that the dissertation titled as “A CLINICAL STUDY ON GENODERMATOSES AND THEIR EFFECT ON QUALITY OF LIFE – PROSPECTIVE OBSERVATIONAL STUDY IN A TERTIARY CARE HOSPITAL” submitted by Dr.P.KARTHIKRAJA to the Tamil Nadu Dr.M.G.R. Medical University, Chennai,in partial fulfilment of the requirement for the award of the Degree of DOCTOR OF MEDICINE in DERMATOLOGY, VENEREOLOGY AND LEPROSY during the academic period 2017 – 2020 is a bonafide research work carried out by him under direct supervision & guidance. Dr.P.Nirmaladevi MD., Dr.S.M.Kannan MS , MCh., Professor and HOD The Dean Department of Dermatology,Venereology &Leprosy Tirunelveli Medical College Tirunelveli Medical College Tirunelveli Tirunelveli CERTIFICATE This is to certify that the dissertation titled as “A CLINICAL STUDY ON GENODERMATOSES AND THEIR EFFECT ON QUALITY OF LIFE – PROSPECTIVE OBSERVATIONAL STUDY IN A TERTIARY CARE HOSPITAL” submitted by Dr.P.KARTHIKRAJA is a original work done by him in the Department of Dermatology,Venereology & Leprosy,Tirunelveli Medical College,Tirunelveli for the award