Useful Commands in Linux and Other Tools for Quality Control
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

At—At, Batch—Execute Commands at a Later Time
at—at, batch—execute commands at a later time at [–csm] [–f script] [–qqueue] time [date] [+ increment] at –l [ job...] at –r job... batch at and batch read commands from standard input to be executed at a later time. at allows you to specify when the commands should be executed, while jobs queued with batch will execute when system load level permits. Executes commands read from stdin or a file at some later time. Unless redirected, the output is mailed to the user. Example A.1 1 at 6:30am Dec 12 < program 2 at noon tomorrow < program 3 at 1945 pm August 9 < program 4 at now + 3 hours < program 5 at 8:30am Jan 4 < program 6 at -r 83883555320.a EXPLANATION 1. At 6:30 in the morning on December 12th, start the job. 2. At noon tomorrow start the job. 3. At 7:45 in the evening on August 9th, start the job. 4. In three hours start the job. 5. At 8:30 in the morning of January 4th, start the job. 6. Removes previously scheduled job 83883555320.a. awk—pattern scanning and processing language awk [ –fprogram–file ] [ –Fc ] [ prog ] [ parameters ] [ filename...] awk scans each input filename for lines that match any of a set of patterns specified in prog. Example A.2 1 awk '{print $1, $2}' file 2 awk '/John/{print $3, $4}' file 3 awk -F: '{print $3}' /etc/passwd 4 date | awk '{print $6}' EXPLANATION 1. Prints the first two fields of file where fields are separated by whitespace. 2. Prints fields 3 and 4 if the pattern John is found. -
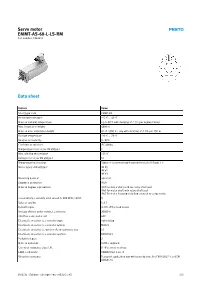
Servo Motor EMMT-AS-60-L-LS-RM Part Number: 5242213
Servo motor EMMT-AS-60-L-LS-RM Part number: 5242213 Data sheet Feature Value Short type code EMMT-AS Ambient temperature -15 °C ... 40 °C Note on ambient temperature Up to 80°C with derating of -1.5% per degree Celsius Max. installation height 4000 m Note on max. installation height As of 1,000 m: only with derating of -1.0% per 100 m Storage temperature -20 °C ... 70 °C Relative air humidity 0 - 90% Conforms to standard IEC 60034 Temperature class as per EN 60034-1 F Max. winding temperature 155 °C Rating class as per EN 60034-1 S1 Temperature monitoring Digital motor temperature transmission via EnDat® 2.2 Motor type to EN 60034-7 IM B5 IM V1 IM V3 Mounting position optional Degree of protection IP40 Note on degree of protection IP40 for motor shaft without rotary shaft seal IP65 for motor shaft with rotary shaft seal IP67 for motor housing including connection components Concentricity, coaxiality, axial runout to DIN SPEC 42955 N Balance quality G 2.5 Detent torque <1.0% of the peak torque Storage lifetime under nominal conditions 20000 h Interface code, motor out 60P Electrical connection 1, connection type Hybrid plug Electrical connection 1, connector system M23x1 Electrical connection 1, number of connections/cores 15 Electrical connection 1, connection pattern 00995913 Pollution degree 2 Note on materials RoHS-compliant Corrosion resistance class CRC 0 - No corrosion stress LABS-Conformity VDMA24364 zone III Vibration resistance Transport application test with severity level 2 to FN 942017-4 and EN 60068-2-6 10/2/21 - Subject to change - Festo AG & Co. -
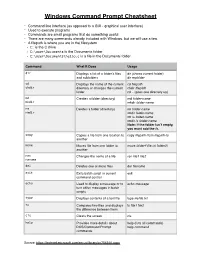
Windows Command Prompt Cheatsheet
Windows Command Prompt Cheatsheet - Command line interface (as opposed to a GUI - graphical user interface) - Used to execute programs - Commands are small programs that do something useful - There are many commands already included with Windows, but we will use a few. - A filepath is where you are in the filesystem • C: is the C drive • C:\user\Documents is the Documents folder • C:\user\Documents\hello.c is a file in the Documents folder Command What it Does Usage dir Displays a list of a folder’s files dir (shows current folder) and subfolders dir myfolder cd Displays the name of the current cd filepath chdir directory or changes the current chdir filepath folder. cd .. (goes one directory up) md Creates a folder (directory) md folder-name mkdir mkdir folder-name rm Deletes a folder (directory) rm folder-name rmdir rmdir folder-name rm /s folder-name rmdir /s folder-name Note: if the folder isn’t empty, you must add the /s. copy Copies a file from one location to copy filepath-from filepath-to another move Moves file from one folder to move folder1\file.txt folder2\ another ren Changes the name of a file ren file1 file2 rename del Deletes one or more files del filename exit Exits batch script or current exit command control echo Used to display a message or to echo message turn off/on messages in batch scripts type Displays contents of a text file type myfile.txt fc Compares two files and displays fc file1 file2 the difference between them cls Clears the screen cls help Provides more details about help (lists all commands) DOS/Command Prompt help command commands Source: https://technet.microsoft.com/en-us/library/cc754340.aspx. -

HEP Computing Part I Intro to UNIX/LINUX Adrian Bevan
HEP Computing Part I Intro to UNIX/LINUX Adrian Bevan Lectures 1,2,3 [email protected] 1 Lecture 1 • Files and directories. • Introduce a number of simple UNIX commands for manipulation of files and directories. • communicating with remote machines [email protected] 2 What is LINUX • LINUX is the operating system (OS) kernel. • Sitting on top of the LINUX OS are a lot of utilities that help you do stuff. • You get a ‘LINUX distribution’ installed on your desktop/laptop. This is a sloppy way of saying you get the OS bundled with lots of useful utilities/applications. • Use LINUX to mean anything from the OS to the distribution we are using. • UNIX is an operating system that is very similar to LINUX (same command names, sometimes slightly different functionalities of commands etc). – There are usually enough subtle differences between LINUX and UNIX versions to keep you on your toes (e.g. Solaris and LINUX) when running applications on multiple platforms …be mindful of this if you use other UNIX flavours. – Mac OS X is based on a UNIX distribution. [email protected] 3 Accessing a machine • You need a user account you should all have one by now • can then log in at the terminal (i.e. sit in front of a machine and type in your user name and password to log in to it). • you can also log in remotely to a machine somewhere else RAL SLAC CERN London FNAL in2p3 [email protected] 4 The command line • A user interfaces with Linux by typing commands into a shell. -

Software Developement Tools Introduction to Software Building
Software Developement Tools Introduction to software building SED & friends Outline 1. an example 2. what is software building? 3. tools 4. about CMake SED & friends – Introduction to software building 2 1 an example SED & friends – Introduction to software building 3 - non-portability: works only on a Unix systems, with mpicc shortcut and MPI libraries and headers installed in standard directories - every build, we compile all files - 0 level: hit the following line: mpicc -Wall -o heat par heat par.c heat.c mat utils.c -lm - 0.1 level: write a script (bash, csh, Zsh, ...) • drawbacks: Example • we want to build the parallel program solving heat equation: SED & friends – Introduction to software building 4 - non-portability: works only on a Unix systems, with mpicc shortcut and MPI libraries and headers installed in standard directories - every build, we compile all files - 0.1 level: write a script (bash, csh, Zsh, ...) • drawbacks: Example • we want to build the parallel program solving heat equation: - 0 level: hit the following line: mpicc -Wall -o heat par heat par.c heat.c mat utils.c -lm SED & friends – Introduction to software building 4 - non-portability: works only on a Unix systems, with mpicc shortcut and MPI libraries and headers installed in standard directories - every build, we compile all files • drawbacks: Example • we want to build the parallel program solving heat equation: - 0 level: hit the following line: mpicc -Wall -o heat par heat par.c heat.c mat utils.c -lm - 0.1 level: write a script (bash, csh, Zsh, ...) SED -

Modern Programming Languages CS508 Virtual University of Pakistan
Modern Programming Languages (CS508) VU Modern Programming Languages CS508 Virtual University of Pakistan Leaders in Education Technology 1 © Copyright Virtual University of Pakistan Modern Programming Languages (CS508) VU TABLE of CONTENTS Course Objectives...........................................................................................................................4 Introduction and Historical Background (Lecture 1-8)..............................................................5 Language Evaluation Criterion.....................................................................................................6 Language Evaluation Criterion...................................................................................................15 An Introduction to SNOBOL (Lecture 9-12).............................................................................32 Ada Programming Language: An Introduction (Lecture 13-17).............................................45 LISP Programming Language: An Introduction (Lecture 18-21)...........................................63 PROLOG - Programming in Logic (Lecture 22-26) .................................................................77 Java Programming Language (Lecture 27-30)..........................................................................92 C# Programming Language (Lecture 31-34) ...........................................................................111 PHP – Personal Home Page PHP: Hypertext Preprocessor (Lecture 35-37)........................129 Modern Programming Languages-JavaScript -

Unix Essentials (Pdf)
Unix Essentials Bingbing Yuan Next Hot Topics: Unix – Beyond Basics (Mon Oct 20th at 1pm) 1 Objectives • Unix Overview • Whitehead Resources • Unix Commands • BaRC Resources • LSF 2 Objectives: Hands-on • Parsing Human Body Index (HBI) array data Goal: Process a large data file to get important information such as genes of interest, sorting expression values, and subset the data for further investigation. 3 Advantages of Unix • Processing files with thousands, or millions, of lines How many reads are in my fastq file? Sort by gene name or expression values • Many programs run on Unix only Command-line tools • Automate repetitive tasks or commands Scripting • Other software, such as Excel, are not able to handle large files efficiently • Open Source 4 Scientific computing resources 5 Shared packages/programs https://tak.wi.mit.edu Request new packages/programs Installed packages/programs 6 Login • Requesting a tak account http://iona.wi.mit.edu/bio/software/unix/bioinfoaccount.php • Windows PuTTY or Cygwin Xming: setup X-windows for graphical display • Macs Access through Terminal 7 Connecting to tak for Windows Command Prompt user@tak ~$ 8 Log in to tak for Mac ssh –Y [email protected] 9 Unix Commands • General syntax Command Options or switches (zero or more) Arguments (zero or more) Example: uniq –c myFile.txt command options arguments Options can be combined ls –l –a or ls –la • Manual (man) page man uniq • One line description whatis ls 10 Unix Directory Structure root / home dev bin nfs lab . jdoe BaRC_Public solexa_public -

101 Useful Linux Commands - Haydenjames.Io
101 Useful Linux Commands - haydenjames.io Some of these commands require elevated permissions (sudo) to run. Enjoy! 1. Execute the previous command used: !! 2. Execute a previous command starting with a specific letter. Example: !s 3. Short way to copy or backup a file before you edit it. For example, copy nginx.conf cp nginx.conf{,.bak} 4. Toggle between current directory and last directory cd - 5. Move to parent (higher level) directory. Note the space! cd .. 6. Go to home directory cd ~ 7. Go to home directory cd $HOME 8. Go to home directory (when used alone) cd 9. Set permissions to 755. Corresponds to these permissions: (-rwx-r-x-r-x), arranged in this sequence: (owner-group-other) chmod 755 <filename> 10. Add execute permission to all users. chmod a+x <filename> 11. Changes ownership of a file or directory to . chown <username> 12. Make a backup copy of a file (named file.backup) cp <file> <file>.backup 13. Copy file1, use it to create file2 cp <file1> <file2> 14. Copy directory1 and all its contents (recursively) into directory2 cp -r <directory1> <directory2>/ 15. Display date date 16. Zero the sdb drive. You may want to use GParted to format the drive afterward. You need elevated permissions to run this (sudo). dd if=/dev/zero of=/dev/sdb 17. Display disk space usage df -h 18. Take detailed messages from OS and input to text file dmesg>dmesg.txt 19. Display a LOT of system information. I usually pipe output to less. You need elevated permissions to run this (sudo). -

Useful Tai Ls Dino
SCIENCE & NATURE Useful Tails Materials Pictures of a possum, horse, lizard, rattlesnake, peacock, fish, bird, and beaver What to do 1. Display the animal pictures so the children can see them. 2. Say the following sentences. Ask the children to guess the animal by the usefulness of its tail. I use my tail for hanging upside down. (possum) I use my tail as a fly swatter. (horse) When my tail breaks off, I grow a new one. (lizard) I shake my noisy tail when I am about to strike. (rattlesnake) My tail opens like a beautiful fan. (peacock) I use my tail as a propeller. I cannot swim without it. (fish) I can’t fly without my tail. (bird) I use my powerful tail for building. (beaver) More to do Ask the children if they can name other animals that have tails. Ask them how these animals’Downloaded tails might by [email protected] useful. from Games: Cut out the tailsProFilePlanner.com of each of the animals. Encourage the children to pin the tails on the pictures (like “Pin the Tail on the Donkey”). Dotti Enderle, Richmond, TX Dino Dig Materials Plastic or rubber dinosaurs or bones Sand Wide-tip, medium-sized paintbrushes Plastic sand shovels Small plastic buckets Clipboards Paper Pencil or pens 508 The GIANT Encyclopedia of Preschool Activities for Four-Year-Olds Downloaded by [email protected] from ProFilePlanner.com SCIENCE & NATURE What to do 1. Beforehand, hide plastic or rubber dinosaurs or bones in the sand. 2. Give each child a paintbrush, shovel, and bucket. 3. -

Your Performance Task Summary Explanation
Lab Report: 11.2.5 Manage Files Your Performance Your Score: 0 of 3 (0%) Pass Status: Not Passed Elapsed Time: 6 seconds Required Score: 100% Task Summary Actions you were required to perform: In Compress the D:\Graphics folderHide Details Set the Compressed attribute Apply the changes to all folders and files In Hide the D:\Finances folder In Set Read-only on filesHide Details Set read-only on 2017report.xlsx Set read-only on 2018report.xlsx Do not set read-only for the 2019report.xlsx file Explanation In this lab, your task is to complete the following: Compress the D:\Graphics folder and all of its contents. Hide the D:\Finances folder. Make the following files Read-only: D:\Finances\2017report.xlsx D:\Finances\2018report.xlsx Complete this lab as follows: 1. Compress a folder as follows: a. From the taskbar, open File Explorer. b. Maximize the window for easier viewing. c. In the left pane, expand This PC. d. Select Data (D:). e. Right-click Graphics and select Properties. f. On the General tab, select Advanced. g. Select Compress contents to save disk space. h. Click OK. i. Click OK. j. Make sure Apply changes to this folder, subfolders and files is selected. k. Click OK. 2. Hide a folder as follows: a. Right-click Finances and select Properties. b. Select Hidden. c. Click OK. 3. Set files to Read-only as follows: a. Double-click Finances to view its contents. b. Right-click 2017report.xlsx and select Properties. c. Select Read-only. d. Click OK. e. -

Unix Tools 2
Unix Tools 2 Jeff Freymueller Outline § Variables as a collec9on of words § Making basic output and input § A sampler of unix tools: remote login, text processing, slicing and dicing § ssh (secure shell – use a remote computer!) § grep (“get regular expression”) § awk (a text processing language) § sed (“stream editor”) § tr (translator) § A smorgasbord of examples Variables as a collec9on of words § The shell treats variables as a collec9on of words § set files = ( file1 file2 file3 ) § This sets the variable files to have 3 “words” § If you want the whole variable, access it with $files § If you want just the second word, use $files[2] § The shell doesn’t count characters, only words Basic output: echo and cat § echo string § Writes a line to standard output containing the text string. This can be one or more words, and can include references to variables. § echo “Opening files” § echo “working on week $week” § echo –n “no carriage return at the end of this” § cat file § Sends the contents of a file to standard output Input, Output, Pipes § Output to file, vs. append to file. § > filename creates or overwrites the file filename § >> filename appends output to file filename § Take input from file, or from “inline input” § < filename take all input from file filename § <<STRING take input from the current file, using all lines un9l you get to the label STRING (see next slide for example) § Use a pipe § date | awk '{print $1}' Example of “inline input” gpsdisp << END • Many programs, especially Okmok2002-2010.disp Okmok2002-2010.vec older ones, interac9vely y prompt you to enter input Okmok2002-2010.gmtvec • You can automate (or self- y Okmok2002-2010.newdisp document) this by using << Okmok2002_mod.stacov • Standard input is set to the 5 contents of this file 5 Okmok2010.stacov between << END and END 5 • You can use any label, not 5 just “END”. -

Bash Crash Course + Bc + Sed + Awk∗
Bash Crash course + bc + sed + awk∗ Andrey Lukyanenko, CSE, Aalto University Fall, 2011 There are many Unix shell programs: bash, sh, csh, tcsh, ksh, etc. The comparison of those can be found on-line 1. We will primary focus on the capabilities of bash v.4 shell2. 1. Each bash script can be considered as a text file which starts with #!/bin/bash. It informs the editor or interpretor which tries to open the file, what to do with the file and how should it be treated. The special character set in the beginning #! is a magic number; check man magic and /usr/share/file/magic on existing magic numbers if interested. 2. Each script (assume you created “scriptname.sh file) can be invoked by command <dir>/scriptname.sh in console, where <dir> is absolute or relative path to the script directory, e.g., ./scriptname.sh for current directory. If it has #! as the first line it will be invoked by this command, otherwise it can be called by command bash <dir>/scriptname.sh. Notice: to call script as ./scriptname.sh it has to be executable, i.e., call command chmod 555 scriptname.sh before- hand. 3. Variable in bash can be treated as integers or strings, depending on their value. There are set of operations and rules available for them. For example: #!/bin/bash var1=123 # Assigns value 123 to var1 echo var1 # Prints ’var1’ to output echo $var1 # Prints ’123’ to output var2 =321 # Error (var2: command not found) var2= 321 # Error (321: command not found) var2=321 # Correct var3=$var2 # Assigns value 321 from var2 to var3 echo $var3 # Prints ’321’ to output