Unix Essentials (Pdf)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

At—At, Batch—Execute Commands at a Later Time
at—at, batch—execute commands at a later time at [–csm] [–f script] [–qqueue] time [date] [+ increment] at –l [ job...] at –r job... batch at and batch read commands from standard input to be executed at a later time. at allows you to specify when the commands should be executed, while jobs queued with batch will execute when system load level permits. Executes commands read from stdin or a file at some later time. Unless redirected, the output is mailed to the user. Example A.1 1 at 6:30am Dec 12 < program 2 at noon tomorrow < program 3 at 1945 pm August 9 < program 4 at now + 3 hours < program 5 at 8:30am Jan 4 < program 6 at -r 83883555320.a EXPLANATION 1. At 6:30 in the morning on December 12th, start the job. 2. At noon tomorrow start the job. 3. At 7:45 in the evening on August 9th, start the job. 4. In three hours start the job. 5. At 8:30 in the morning of January 4th, start the job. 6. Removes previously scheduled job 83883555320.a. awk—pattern scanning and processing language awk [ –fprogram–file ] [ –Fc ] [ prog ] [ parameters ] [ filename...] awk scans each input filename for lines that match any of a set of patterns specified in prog. Example A.2 1 awk '{print $1, $2}' file 2 awk '/John/{print $3, $4}' file 3 awk -F: '{print $3}' /etc/passwd 4 date | awk '{print $6}' EXPLANATION 1. Prints the first two fields of file where fields are separated by whitespace. 2. Prints fields 3 and 4 if the pattern John is found. -

ECOGEO Workshop 2: Introduction to Env 'Omics
ECOGEO Workshop 2: Introduction to Env ‘Omics Unix and Bioinformatics Ben Tully (USC); Ken Youens-Clark (UA) Unix Commands pwd rm grep tail install ls ‘>’ sed cut cd cat nano top mkdir ‘<’ history screen touch ‘|’ $PATH ssh cp sort less df mv uniq head rsync/scp Unix Command Line 1. Open Terminal window Unix Command Line 2. Open Chrome and navigate to Unix tutorial at Protocols.io 3. Group: ECOGEO 4. Protocol: ECOGEO Workshop 2: Unix Module ! This will allow you to copy, paste Unix scripts into terminal window ! ECOGEO Protocols.io for making copy, paste easier Unix Command Line $ ls ls - lists items in the current directory Many commands have additional options that can be set by a ‘-’ $ ls -a Unix Command Line $ ls -a lists all files/directories, including hidden files ‘.’ $ ls -l lists the long format File Permissions | # Link | User | Group | Size | Last modified $ ls -lt lists the long format, but ordered by date last modified Unix Command Line Unix Command Line $ cd ecogeo/ cd - change directory List the contents of the current directory Move into the directory called unix List contents $ pwd pwd - present working directory Unix Command Line /home/c-debi/ecogeo/unix When were we in the directory home? Or c-debi? Or ecogeo? $ cd / Navigates to root directory List contents of root directory This where everything is stored in the computer All the commands we are running live in /bin Unix Command Line / root bin sys home mnt usr c-debi BioinfPrograms cdebi Desktop Downloads ecogeo unix assembly annotation etc Typical Unix Layout Unix Command Line Change directory to home Change directory to c-debi Change directory to ecogeo Change directory to unix List contents Change directory to data Change directory to root Unix Command Line Change directory to unix/data in one step $ cd /home/c-debi/ecogeo/unix/data Tab can be used to auto complete names $ cd . -

Introduction to Unix
Introduction to Unix Rob Funk <[email protected]> University Technology Services Workstation Support http://wks.uts.ohio-state.edu/ University Technology Services Course Objectives • basic background in Unix structure • knowledge of getting started • directory navigation and control • file maintenance and display commands • shells • Unix features • text processing University Technology Services Course Objectives Useful commands • working with files • system resources • printing • vi editor University Technology Services In the Introduction to UNIX document 3 • shell programming • Unix command summary tables • short Unix bibliography (also see web site) We will not, however, be covering these topics in the lecture. Numbers on slides indicate page number in book. University Technology Services History of Unix 7–8 1960s multics project (MIT, GE, AT&T) 1970s AT&T Bell Labs 1970s/80s UC Berkeley 1980s DOS imitated many Unix ideas Commercial Unix fragmentation GNU Project 1990s Linux now Unix is widespread and available from many sources, both free and commercial University Technology Services Unix Systems 7–8 SunOS/Solaris Sun Microsystems Digital Unix (Tru64) Digital/Compaq HP-UX Hewlett Packard Irix SGI UNICOS Cray NetBSD, FreeBSD UC Berkeley / the Net Linux Linus Torvalds / the Net University Technology Services Unix Philosophy • Multiuser / Multitasking • Toolbox approach • Flexibility / Freedom • Conciseness • Everything is a file • File system has places, processes have life • Designed by programmers for programmers University Technology Services -

Useful Commands in Linux and Other Tools for Quality Control
Useful commands in Linux and other tools for quality control Ignacio Aguilar INIA Uruguay 05-2018 Unix Basic Commands pwd show working directory ls list files in working directory ll as before but with more information mkdir d make a directory d cd d change to directory d Copy and moving commands To copy file cp /home/user/is . To copy file directory cp –r /home/folder . to move file aa into bb in folder test mv aa ./test/bb To delete rm yy delete the file yy rm –r xx delete the folder xx Redirections & pipe Redirection useful to read/write from file !! aa < bb program aa reads from file bb blupf90 < in aa > bb program aa write in file bb blupf90 < in > log Redirections & pipe “|” similar to redirection but instead to write to a file, passes content as input to other command tee copy standard input to standard output and save in a file echo copy stream to standard output Example: program blupf90 reads name of parameter file and writes output in terminal and in file log echo par.b90 | blupf90 | tee blup.log Other popular commands head file print first 10 lines list file page-by-page tail file print last 10 lines less file list file line-by-line or page-by-page wc –l file count lines grep text file find lines that contains text cat file1 fiel2 concatenate files sort sort file cut cuts specific columns join join lines of two files on specific columns paste paste lines of two file expand replace TAB with spaces uniq retain unique lines on a sorted file head / tail $ head pedigree.txt 1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 0 8 0 0 9 0 0 10 -

Install and Run External Command Line Softwares
job monitor and control top: similar to windows task manager (space to refresh, q to exit) w: who is there ps: all running processes, PID, status, type ps -ef | grep yyin bg: move current process to background fg: move current process to foreground jobs: list running and suspended processes kill: kill processes kill pid (could find out using top or ps) 1 sort, cut, uniq, join, paste, sed, grep, awk, wc, diff, comm, cat All types of bioinformatics sequence analyses are essentially text processing. Unix Shell has the above commands that are very useful for processing texts and also allows the output from one command to be passed to another command as input using pipe (“|”). less cosmicRaw.txt | cut -f2,3,4,5,8,13 | awk '$5==22' | cut -f1 | sort -u | wc This makes the processing of files using Shell very convenient and very powerful: you do not need to write output to intermediate files or load all data into the memory. For example, combining different Unix commands for text processing is like passing an item through a manufacturing pipeline when you only care about the final product 2 Hands on example 1: cosmic mutation data - Go to UCSC genome browser website: http://genome.ucsc.edu/ - On the left, find the Downloads link - Click on Human - Click on Annotation database - Ctrl+f and then search “cosmic” - On “cosmic.txt.gz” right-click -> copy link address - Go to the terminal and wget the above link (middle click or Shift+Insert to paste what you copied) - Similarly, download the “cosmicRaw.txt.gz” file - Under your home, create a folder -

CS2043 - Unix Tools & Scripting Cornell University, Spring 20141
CS2043 - Unix Tools & Scripting Cornell University, Spring 20141 Instructor: Bruno Abrahao January 31, 2014 1 Slides evolved from previous versions by Hussam Abu-Libdeh and David Slater Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Vim: Tip of the day! Line numbers Displays line number in Vim: :set nu Hides line number in Vim: :set nonu Goes to line number: :line number Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Counting wc How many lines of code are in my new awesome program? How many words are in this document? Good for bragging rights Word, Character, Line, and Byte count with wc wc -l : count the number of lines wc -w : count the number of words wc -m : count the number of characters wc -c : count the number of bytes Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Sorting sort Sorts the lines of a text file alphabetically. sort -ru file sorts the file in reverse order and deletes duplicate lines. sort -n -k 2 -t : file sorts the file numerically by using the second column, separated by a colon Example Consider a file (numbers.txt) with the numbers 1, 5, 8, 11, 62 each on a separate line, then: $ sort numbers.txt $ sort numbers.txt -n 1 1 11 5 5 8 62 11 8 62 Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting uniq uniq uniq file - Discards all but one of successive identical lines uniq -c file - Prints the number of successive identical lines next to each line Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Character manipulation! The Translate Command tr [options] <char_list1> [char_list2] Translate or delete characters char lists are strings of characters By default, searches for characters in char list1 and replaces them with the ones that occupy the same position in char list2 Example: tr 'AEIOU' 'aeiou' - changes all capital vowels to lower case vowels Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Pipes and redirection tr only receives input from standard input (stdin) i.e. -

IBM Education Assistance for Z/OS V2R1
IBM Education Assistance for z/OS V2R1 Item: ASCII Unicode Option Element/Component: UNIX Shells and Utilities (S&U) Material is current as of June 2013 © 2013 IBM Corporation Filename: zOS V2R1 USS S&U ASCII Unicode Option Agenda ■ Trademarks ■ Presentation Objectives ■ Overview ■ Usage & Invocation ■ Migration & Coexistence Considerations ■ Presentation Summary ■ Appendix Page 2 of 19 © 2013 IBM Corporation Filename: zOS V2R1 USS S&U ASCII Unicode Option IBM Presentation Template Full Version Trademarks ■ See url http://www.ibm.com/legal/copytrade.shtml for a list of trademarks. Page 3 of 19 © 2013 IBM Corporation Filename: zOS V2R1 USS S&U ASCII Unicode Option IBM Presentation Template Full Presentation Objectives ■ Introduce the features and benefits of the new z/OS UNIX Shells and Utilities (S&U) support for working with ASCII/Unicode files. Page 4 of 19 © 2013 IBM Corporation Filename: zOS V2R1 USS S&U ASCII Unicode Option IBM Presentation Template Full Version Overview ■ Problem Statement –As a z/OS UNIX Shells & Utilities user, I want the ability to control the text conversion of input files used by the S&U commands. –As a z/OS UNIX Shells & Utilities user, I want the ability to run tagged shell scripts (tcsh scripts and SBCS sh scripts) under different SBCS locales. ■ Solution –Add –W filecodeset=codeset,pgmcodeset=codeset option on several S&U commands to enable text conversion – consistent with support added to vi and ex in V1R13. –Add –B option on several S&U commands to disable automatic text conversion – consistent with other commands that already have this override support. –Add new _TEXT_CONV environment variable to enable or disable text conversion. -

User's Manual
USER’S MANUAL USER’S > UniQTM UniQTM User’s Manual Ed.: 821003146 rev.H © 2016 Datalogic S.p.A. and its Group companies • All rights reserved. • Protected to the fullest extent under U.S. and international laws. • Copying or altering of this document is prohibited without express written consent from Datalogic S.p.A. • Datalogic and the Datalogic logo are registered trademarks of Datalogic S.p.A. in many countries, including the U.S. and the E.U. All other brand and product names mentioned herein are for identification purposes only and may be trademarks or registered trademarks of their respective owners. Datalogic shall not be liable for technical or editorial errors or omissions contained herein, nor for incidental or consequential damages resulting from the use of this material. Printed in Donnas (AO), Italy. ii SYMBOLS Symbols used in this manual along with their meaning are shown below. Symbols and signs are repeated within the chapters and/or sections and have the following meaning: Generic Warning: This symbol indicates the need to read the manual carefully or the necessity of an important maneuver or maintenance operation. Electricity Warning: This symbol indicates dangerous voltage associated with the laser product, or powerful enough to constitute an electrical risk. This symbol may also appear on the marking system at the risk area. Laser Warning: This symbol indicates the danger of exposure to visible or invisible laser radiation. This symbol may also appear on the marking system at the risk area. Fire Warning: This symbol indicates the danger of a fire when processing flammable materials. -
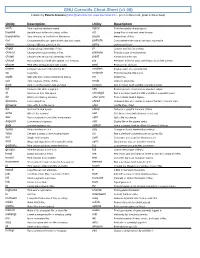
GNU Coreutils Cheat Sheet (V1.00) Created by Peteris Krumins ([email protected], -- Good Coders Code, Great Coders Reuse)
GNU Coreutils Cheat Sheet (v1.00) Created by Peteris Krumins ([email protected], www.catonmat.net -- good coders code, great coders reuse) Utility Description Utility Description arch Print machine hardware name nproc Print the number of processors base64 Base64 encode/decode strings or files od Dump files in octal and other formats basename Strip directory and suffix from file names paste Merge lines of files cat Concatenate files and print on the standard output pathchk Check whether file names are valid or portable chcon Change SELinux context of file pinky Lightweight finger chgrp Change group ownership of files pr Convert text files for printing chmod Change permission modes of files printenv Print all or part of environment chown Change user and group ownership of files printf Format and print data chroot Run command or shell with special root directory ptx Permuted index for GNU, with keywords in their context cksum Print CRC checksum and byte counts pwd Print current directory comm Compare two sorted files line by line readlink Display value of a symbolic link cp Copy files realpath Print the resolved file name csplit Split a file into context-determined pieces rm Delete files cut Remove parts of lines of files rmdir Remove directories date Print or set the system date and time runcon Run command with specified security context dd Convert a file while copying it seq Print sequence of numbers to standard output df Summarize free disk space setuidgid Run a command with the UID and GID of a specified user dir Briefly list directory -

07 07 Unixintropart2 Lucio Week 3
Unix Basics Command line tools Daniel Lucio Overview • Where to use it? • Command syntax • What are commands? • Where to get help? • Standard streams(stdin, stdout, stderr) • Pipelines (Power of combining commands) • Redirection • More Information Introduction to Unix Where to use it? • Login to a Unix system like ’kraken’ or any other NICS/ UT/XSEDE resource. • Download and boot from a Linux LiveCD either from a CD/DVD or USB drive. • http://www.puppylinux.com/ • http://www.knopper.net/knoppix/index-en.html • http://www.ubuntu.com/ Introduction to Unix Where to use it? • Install Cygwin: a collection of tools which provide a Linux look and feel environment for Windows. • http://cygwin.com/index.html • https://newton.utk.edu/bin/view/Main/Workshop0InstallingCygwin • Online terminal emulator • http://bellard.org/jslinux/ • http://cb.vu/ • http://simpleshell.com/ Introduction to Unix Command syntax $ command [<options>] [<file> | <argument> ...] Example: cp [-R [-H | -L | -P]] [-fi | -n] [-apvX] source_file target_file Introduction to Unix What are commands? • An executable program (date) • A command built into the shell itself (cd) • A shell program/function • An alias Introduction to Unix Bash commands (Linux) alias! crontab! false! if! mknod! ram! strace! unshar! apropos! csplit! fdformat! ifconfig! more! rcp! su! until! apt-get! cut! fdisk! ifdown! mount! read! sudo! uptime! aptitude! date! fg! ifup! mtools! readarray! sum! useradd! aspell! dc! fgrep! import! mtr! readonly! suspend! userdel! awk! dd! file! install! mv! reboot! symlink! -

Cloud Computing and Unix: an Introduction
Cloud Computing and Unix: An Introduction Dr. Sophie Shaw University of Aberdeen, UK [email protected] Aberdeen London Exeter What We’re Going To Do • Why Unix? • Cloud Computing • Connecting to AWS • Introduction to Unix Commands Etiquette • PowerPoint interspersed with Challenges • Ask me questions • Ask demonstrators • Work together • Cheat! What is Unix? • Operating System Why Unix? • Bioinformatics software designed to run on Unix platforms. • Large amounts of data. • Much faster than your Windows PC. How Can We Use Unix? • Linux computers or servers. • Compute clusters. • The cloud. – What we’re going to use this week So What is Cloud Computing? Cloud Computing Solutions AWS “Availability Zones” and Data Centres How it Works AMI (“Amazon Machine Image”) Base computer with all data and software How it Works Own copy of the AMI = Instance (Virtual Machine or VM) Terminology • Creating an instance – buying a brand new computer with software already installed. • Starting an instance – turning that computer on. • Stopping an instance – turning that computer off. • Terminating an instance – setting that computer on fire and throwing it out of the window. The Rules • Only create one instance each. • Stop your instance at the end of each day (unless you have software running). • Name your instance (with YOUR name! No Bruce Waynes please) • Only start or stop your own instance. • Only terminate your own instance. Connecting to Your Instance Remote Desktop Secure Shell – Software “SSH” e.g. X2Go e.g. SSH or PuTTY Now What?! • You’re each going -

How to Build a Search-Engine with Common Unix-Tools
The Tenth International Conference on Advances in Databases, Knowledge, and Data Applications Mai 20 - 24, 2018 - Nice/France How to build a Search-Engine with Common Unix-Tools Andreas Schmidt (1) (2) Department of Informatics and Institute for Automation and Applied Informatics Business Information Systems Karlsruhe Institute of Technologie University of Applied Sciences Karlsruhe Germany Germany Andreas Schmidt DBKDA - 2018 1/66 Resources available http://www.smiffy.de/dbkda-2018/ 1 • Slideset • Exercises • Command refcard 1. all materials copyright, 2018 by andreas schmidt Andreas Schmidt DBKDA - 2018 2/66 Outlook • General Architecture of an IR-System • Naive Search + 2 hands on exercices • Boolean Search • Text analytics • Vector Space Model • Building an Inverted Index & • Inverted Index Query processing • Query Processing • Overview of useful Unix Tools • Implementation Aspects • Summary Andreas Schmidt DBKDA - 2018 3/66 What is Information Retrieval ? Information Retrieval (IR) is finding material (usually documents) of an unstructured nature (usually text) that satisfies an informa- tion need (usually a query) from within large collections (usually stored on computers). [Manning et al., 2008] Andreas Schmidt DBKDA - 2018 4/66 What is Information Retrieval ? need for query information representation how to match? document document collection representation Andreas Schmidt DBKDA - 2018 5/66 Keyword Search • Given: • Number of Keywords • Document collection • Result: • All documents in the collection, cotaining the keywords • (ranked by relevance) Andreas Schmidt DBKDA - 2018 6/66 Naive Approach • Iterate over all documents d in document collection • For each document d, iterate all words w and check, if all the given keywords appear in this document • if yes, add document to result set • Output result set • Extensions/Variants • Ranking see examples later ...