Proteomics of Serum Extracellular Vesicles Identifies a Novel COPD Biomarker, Fibulin-3 from Elastic Fibres
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
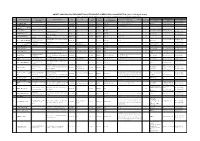
As of 15Th.April.2020) *Items Listed May Be Modified Along with the Circumstances of Supervising Organizations
ADDITIONAL ENGLISH INFORMATION OF EXCELLENT SUPERVISING ORGANIZATION (As of 15th.April.2020) *Items listed may be modified along with the circumstances of supervising organizations. In that case, we will modify the list shortly. Job Categories (Eligible when Interns shifts to No. of Person in charge Name of Organization Date of Approval Date of Expiry Accepting Country No. Name of Organization Address in English TEL HP Technical Intern Training (ii)) *Refer to Implementing TEL (in English) (Y/M/D) (Y/M/D) *Refer to Country Code Name Email Category Code Organizations* FAX ISS HOKKAIDO business 〒067-0056, 2F No.225 Mihara, Ebetsu-shi, TEL: 011-807-0491 1 ISS北海道事業協同組合 0118070491 N/A 2018/10/31 2023/10/30 CHN, VNM 1-1, 1-2, 3-21, 4-2, 4-9, 4-10, 7-6 70 佐藤(Sato) [email protected] cooperative association Hokkaido FAX: 011-807-0492 2 網走国際交流協同組合 0152462447 2018/12/10 2023/12/9 CHN 1-2, 4-2, 4-4, 4-6 7 アパレルファッション研究協同組合 3 0139551525 2018/1/22 2023/1/21 KHM 5-6 4 (事業休止中) 〒058-0204, 182-2 Erimocho Honchou, TEL: 01466-2-2211 4 えりも漁業協同組合 Erimo Fishery Cooperative 0146622211 http://www.jf-erimo.or.jp 2018/9/28 2023/9/27 IDN, VNM 2-1 5 岡道(Okamichi)(日本国内) [email protected] Horoizumi-gun, Hokkaido FAX: 01466-2-4090 〒049-2222, 2-188-1 Sawara, Mori-machi, [email protected] TEL: 01374-8-5111 5 渡島国際交流事業協同組合 N/A 0137485111 N/A 2017/11/1 2022/10/31 CHN, VNM 4-3, 4-4 18 谷垣 (TANIGAKI) Kayabe-gun, Hokkaido .jp FAX: 01374-8-5112 Okhotsk International Business 〒094-0002, 1-2-42 Masago-cho, Monbetsu-shi, 6 オホーツク国際事業協同組合 0158236686 N/A 2018/6/29 2023/6/28 CHN, THA 4-3, 4-4, 4-5 18 Association Hokkaido OKHOTSK INTERNATIONAL 〒098-1702, 91-5 Oumu, Oumu-cho, Monbetsu- [email protected]. -

2018 JSAE Annual Congress (Spring)
22001188 JSAJSAEE AnAnnnualual CongressCongress ((SpringSpring)) Wednesday, May 23 - Friday, May 25 2018 / Pacifico Yokohama Final Program Floor Map Customer Service EV EV EV EV EV Inter Continental Yokohama Grand Annex Exhibition Hall Hall May 23, 24メインホール Forum May 24 Keynote Address Conference Center セルフ On-site Registration ビジネスセンター EV EV Beverages Book Store Print Service EV EV EV EV EV Workshop 2F Harbor Lounge (Please proceed from the Exhibition Hall) Help Desk ① ② Forum ③ ④ Registration ⑤ ⑥ Registration Automotive Engineering Exposition 2018 ⑦ ⑧ Pre-registered Attendee ⑨ ⑩ ⑪ ⑫ Lecture/ Special Lecture EV EV Room 419 Tea Room Room Room 315 418 Presentation Room Presentation Room Room Presentation Room Room 417 314 + + 416 313 Room May 24 Awards Ceremony Room 312 415 + JSAE Annual Meeting + 311 414 Room 512 トイレ使用不可 Beverages + Room 511 502 Room May 24 JSAE Annual Room 503 Room Room Party 413 Room 302 304 501 Room Room Room 412 Room Registration 411 Chairperson 301 303 YOKOHAMA Booth No.98 NAGOYA Booth No.186 Speaker Room Room Room 316 317 318 Cloak Room Presentation Preparation Room Smoking Area Beverages Smoking Area 春季大会プログラム英文..indd 1 18/04/04 17:01 2018 JSAE Annual Congress (Spring) Period : Wednesday, May 23 to Friday, May 25, 2018 Venue : PACIFICO YOKOHAMA Table of Contents Timetable Wednesday, May 23 …………………………………………… 2,3 Thursday, May 24 ………………………………………………… 4,5 Friday, May 25 …………………………………………………… 6,7 Information …………………………………………………………………… 8,9 Events………………………………………………………………………… 10,11 Forum ………………………………………………………………………… 12-20 -

Subtype-Specific Gout Susceptibility Loci and Enrichment of Selection
Ann Rheum Dis: first published as 10.1136/annrheumdis-2019-216644 on 1 April 2020. Downloaded from Crystal arthropathies EPIDEMIOLOGICAL SCIENCE Subtype- specific gout susceptibility loci and enrichment of selection pressure on ABCG2 and ALDH2 identified by subtype genome- wide meta- analyses of clinically defined gout patients Akiyoshi Nakayama,1,2 Masahiro Nakatochi ,3 Yusuke Kawamura,1,4 Ken Yamamoto,5 Hirofumi Nakaoka,6 Seiko Shimizu,1 Toshihide Higashino,1,7 Teruhide Koyama,8 Asahi Hishida,9 Kiyonori Kuriki,10 Miki Watanabe,11 Toru Shimizu,12,13 Keiko Ooyama,14 Hiroshi Ooyama,14 Mitsuo Nagase,15 Yuji Hidaka,16 Daisuke Matsui,8 Takashi Tamura,9 Takeshi Nishiyama,11 Chisato Shimanoe,17,18 Sakurako Katsuura- Kamano,19 Naoyuki Takashima,20,21 Yuya Shirai,22,23 Makoto Kawaguchi,1,24 Mikiya Takao,1,25 Ryo Sugiyama,1 Yuzo Takada,26 Takahiro Nakamura,27 Hiroshi Nakashima,28 Masashi Tsunoda,28 Inaho Danjoh,29 Atsushi Hozawa,30 Kazuyoshi Hosomichi,31 Yu Toyoda,32 Yu Kubota,32 Tappei Takada,32 Hiroshi Suzuki,32 Blanka Stiburkova,33,34 Tanya J. Major,35 Tony R. Merriman,35 Nagato Kuriyama,8 Haruo Mikami,36 Toshiro Takezaki,37 Keitaro Matsuo,38,39 Sadao Suzuki,11 Tatsuo Hosoya,40,41 Yoichiro Kamatani,42,43 Michiaki Kubo,44 Kimiyoshi Ichida,40,45 Kenji Wakai,9 Ituro Inoue,6 Yukinori Okada ,22,46 Nariyoshi Shinomiya,1 Hirotaka Matsuo,1 on behalf of Japan Gout Genomics Consortium (Japan Gout) copyright. Handling editor Josef S Smolen ABSTRact Key messages ► Additional material is Objectives Genome- wide meta- analyses of clinically published online only. To view defined gout were performed to identify subtype- please visit the journal online What is already known about this subject? (http:// dx. -

COVID-19 and Rheumatology
Annals of the Rheumatic Diseases publishes original work on all aspects of rheumatology ARDThe EULAR Journal Editor Josef S Smolen (Austria) and disorders of connective tissue. Laboratory Associate Editors Francis Berenbaum (France) and clinical studies are equally welcome Dimitrios Boumpas (Greece) Gerd Burmester (Germany) Editorial Board Mary Crow (USA) Daniel Aletaha (Austria) Rik Lories (Belgium) Kimme Hyrich (UK) Johan Askling (Sweden) Ingrid Lundberg (Sweden) Contact Details Rik Lories (Belgium) Sang-Cheol Bae (Korea) Gary MacFarlane (UK) Iain McInnes (UK) Xenofon Baraliakos (Germany) Xavier Mariette (France) Editorial Office Thomas Pap (Germany) Anne Barton (UK) Alberto Martini (Italy) Annals of the Rheumatic Diseases David Pisetsky (USA) Maarten Boers (The Netherlands) Marco Mattuci Cerinic (Italy) BMJ Journals, BMA House, Tavistock Square Désirée van der Heijde Maxine Breban (France) Dennis McGonagle (UK) London WCIH 9JR,UK (The Netherlands) Matthew Brown (Australia) Fred Miller (USA) Kazuhiko Yamamoto (Japan) Maya Buch (UK) Peter Nash (Australia) E: [email protected] Loreto Carmona (Spain) Michael Nurmohamed (The Methodological and Statistical Carlo Chizzolini (Switzerland) Netherlands) Production Editor Advisor Bernard Combe (France) Caroline Ospelt (Switzerland) Teresa Jobson Philip Conaghan (UK) Stian Lydersen (Norway) Monika Østensen (Norway) E: [email protected] Maurizio Cutolo (Italy) Constatino Pitzalis (UK) Social Media Advisors Nicola Dalbeth (Australia) Alessia Alunno (Italy) Jane Salmon (USA) EULAR Christian Dejaco -

All-Time Great Remy Bonjasky Makes Successful Return to the Ring, Defeats Anderson ‘Braddock’ Silva in Hard- Hitting Heavyweight Main Event
ALL-TIME GREAT REMY BONJASKY MAKES SUCCESSFUL RETURN TO THE RING, DEFEATS ANDERSON ‘BRADDOCK’ SILVA IN HARD- HITTING HEAVYWEIGHT MAIN EVENT BRUSSELS, Belgium (Oct. 6, 2012) – Three years away from the ring. Three years in which Remy Bonjasky had grown older while the new generation of prospects ripened and matured into the contenders of today. The question hanging over this fight was whether Bonjasky can hang with the prime young men of the heavyweight division – and the answer was “Yes.” Bonjasky hadn’t been offered an easy comeback either. GLORY had welcomed him with open arms but only if he was willing to test himself against a rising star and prove that he could be competitive in what is by far the kickboxing world’s toughest and most stacked division. And so Bonjasky found himself opposite Anderson ‘Braddock’ Silva, the man who nearly beat the very dangerous Badr Hari earlier this year, at Vorst Nationaal Arena. Braddock’s name may not be as big as Bonjasky’s but, make no mistake, he is not someone who puts a lot of store in fame. A win over Bonjasky would have been enormous for his career and so he put his all into the bout. The pace was incredible from opening seconds, with both men hammering low kicks into each other repeatedly as they sought to slow the opposing man down. There were extended leg-kick trades several times as the pair went back and forth, one for one, until one of them did something to break the cycle. But by the mid-point of round two it was plain to see that they were each feeling the hurt from the strikes they had taken on the thighs, strikes which would have rendered a normal man unable to walk. -

Program May 14 (Thursday) Presidential Lecturepresidential
Plenary Program May 14 (Thursday) Presidential Lecture 16:20-17:05 Plenary Lecture 01 Room: National Convention Hall Chairperson: Teruo Kawada (Kyoto University, Japan) PL01 Leptin and the Regulation of Food Intake and Body Weight Jeffrey M. Friedman Rockefeller University, USA Educational 17:05-17:50 Plenary Lecture 02 Room: National Convention Hall Chairperson: Tohru Fushiki (Ryukoku University, Japan) PL02 Functional Food Science in Japan: Present State and Perspectives Keiko Abe Symposium The University of Tokyo, Japan, the Kanagawa Academy of Science & Technology (KAST), Japan May 15 (Friday) 9:00-9:45 Plenary Lecture 03 Room: Main Hall at Conference Center (Satellite Viewing is available in Room 301-304) Sponsored Symposium Chairperson: Chizuru Nishida (World Health Organization (WHO), Switzerland) PL03 The Present Role of Industrial Food Processing in Food Systems and Its Implications for Controlling the Obesity Pandemic Carlos A. Monteiro University of São Paulo, Brazil May 16 (Saturday) Luncheon 9:00-9:45 Plenary Lecture 04 Room: Main Hall at Conference Center (Satellite Viewing is available in Room 301-304) Chairperson: Pek-Yee Chow (Federation of Asian Nutrition Societies, Singapore Nutrition and Dietetics Association (SNDA), Singapore) PL04 Multi-Stakeholders and Multi-Strategic Approaches for Food and Nutrition Security Evening Kraisid Tontisirin Mahidol University, Thailand May 17 (Sunday) FANS Report FANS 9:00-9:45 Plenary Lecture 05 Room: Main Hall at Conference Center (Satellite Viewing is available in Room 301-304) Chairperson: -

Subtype-Specific Gout Susceptibility Loci and Enrichment of Selection
Crystal arthropathies Ann Rheum Dis: first published as 10.1136/annrheumdis-2019-216644 on 1 April 2020. Downloaded from EPIDEMIOLOGICAL SCIENCE Subtype- specific gout susceptibility loci and enrichment of selection pressure on ABCG2 and ALDH2 identified by subtype genome- wide meta- analyses of clinically defined gout patients Akiyoshi Nakayama,1,2 Masahiro Nakatochi ,3 Yusuke Kawamura,1,4 Ken Yamamoto,5 Hirofumi Nakaoka,6 Seiko Shimizu,1 Toshihide Higashino,1,7 Teruhide Koyama,8 Asahi Hishida,9 Kiyonori Kuriki,10 Miki Watanabe,11 Toru Shimizu,12,13 Keiko Ooyama,14 Hiroshi Ooyama,14 Mitsuo Nagase,15 Yuji Hidaka,16 Daisuke Matsui,8 Takashi Tamura,9 Takeshi Nishiyama,11 Chisato Shimanoe,17,18 Sakurako Katsuura- Kamano,19 Naoyuki Takashima,20,21 Yuya Shirai,22,23 Makoto Kawaguchi,1,24 Mikiya Takao,1,25 Ryo Sugiyama,1 Yuzo Takada,26 Takahiro Nakamura,27 Hiroshi Nakashima,28 Masashi Tsunoda,28 Inaho Danjoh,29 Atsushi Hozawa,30 Kazuyoshi Hosomichi,31 Yu Toyoda,32 Yu Kubota,32 Tappei Takada,32 Hiroshi Suzuki,32 Blanka Stiburkova,33,34 Tanya J. Major,35 Tony R. Merriman,35 Nagato Kuriyama,8 Haruo Mikami,36 Toshiro Takezaki,37 Keitaro Matsuo,38,39 Sadao Suzuki,11 Tatsuo Hosoya,40,41 Yoichiro Kamatani,42,43 Michiaki Kubo,44 Kimiyoshi Ichida,40,45 Kenji Wakai,9 Ituro Inoue,6 Yukinori Okada ,22,46 Nariyoshi Shinomiya,1 Hirotaka Matsuo,1 on behalf of Japan Gout Genomics Consortium (Japan Gout) Handling editor Josef S Smolen ABSTRact Key messages ► Additional material is Objectives Genome- wide meta- analyses of clinically published online only. To view defined gout were performed to identify subtype- please visit the journal online What is already known about this subject? (http:// dx. -
Annual Autumn Meeting of JASNAOE 2017 Program
Annual Autumn Meeting of JASNAOE 2017 Program Date: 27th (Mon.), 28th (Tue.) November, 2017 Place: Internatinal Conference Center Hiroshima 27th November (Monday) Time Title Author(s) (〇Speaker) 【Conference Room 1】 9:20 ~ 10:20 General Session (CFD1) Chair: Yasuyuki Toda (Osaka Univ.) 9:20 ~ 9:40 Validation of Full-Scale Ship CFD Simulation by the Direct Comparison with Sea Trials ○Natsumi Murakami, Takanori Hino Results 9:40 ~ 10:00 Viscous CFD Analysis of Working Principle for Pre-Swirl Stator Fins Equipped on a ○Nobuaki SAKAMOTO, Kenji MOKUO, Masahiro Tanker TAMASHIMA 10:00 ~ 10:20 Forces, Ship Motions and Nominal Wake for KCS Ship Model in Regular Head Waves ○Ping-Chen Wu, Md Alfaz Hossain, Htike Aung Kyaw, Ayaka Matsumoto, Yasuyuki Toda 10:40 ~ 12:00 General Session (Ship Design) Chair: Yusuke Tahara (NMRI) 10:40 ~ 11:00 Study on a Hull Optimization Using Swarm Intelligence Tomoaki Egami, ○Akihito Hirayama 11:00 ~ 11:20 Development of a Small Stick-Held Dip Net Fishing Boat ○Toshihiko Kawashima, Hitoshi Nishimoto, Yasuaki Doi, Hidemi Mutsuda, Hideki Matsuda 11:20 ~ 11:40 Hull Improvement of a Fixed Shore Net Fishing Boat ○Toshihiko Kawashima, Hitoshi Nishimoto, Yasuaki Doi, Hidemi Mutsuda, Hideki Matsuda 11:40 ~ 12:00 Development of Automatic Hull Form Generation Method to Yield Specific Wake Field. ○Yasuo Ichinose, Yusuke Tahara 13:00 ~ 13:20 Poster Session : One-minute presentation 13:20 ~ 14:40 General Session (Propulsion Performance 1) Chair: Chiharu Kawakita (NMRI) 13:20 ~ 13:40 Performance confirmation and application of improved -

Association of the RPA3-UMAD1 Locus with Interstitial Lung Diseases
Rheumatoid arthritis Ann Rheum Dis: first published as 10.1136/annrheumdis-2020-217256 on 31 July 2020. Downloaded from TRANSLATIONAL SCIENCE Association of the RPA3- UMAD1 locus with interstitial lung diseases complicated with rheumatoid arthritis in Japanese Yuya Shirai,1,2 Suguru Honda,3 Katsunori Ikari,3 Masahiro Kanai,1,4 Yoshito Takeda,2 Yoichiro Kamatani,5,6 Takayuki Morisaki,7,8 Eiichi Tanaka,3 Atsushi Kumanogoh,2,9,10 Masayoshi Harigai ,3 Yukinori Okada 1,6,10,11 Handling editor Josef S ABSTRACT Key messages Smolen Objectives The genetic background of rheumatoid arthritis–interstitial lung disease (RA-ILD) has been ► Additional material is What is already known about this subject? published online only. To view evaluated in Europeans, but little knowledge has been ► The genetic background of rheumatoid please visit the journal online obtained in non- Europeans. This study aimed to elucidate arthritis–interstitial lung disease (RA- ILD) (http:// dx. doi. org/ 10. 1136/ genome- wide risk of RA- ILD in non- Europeans. has been evaluated in Europeans, but annrheumdis- 2020- 217256). Methods We performed an initial genome-wide little knowledge has been obtained in association study (GWAS) of RA-ILD in the Japanese For numbered affiliations see non- Europeans. end of article. population. By conducting the meta-analysis of the The genetic background of RA- ILD has not yet three GWAS datasets of the RA cohorts and biobank ► been evaluated in the context of genome-wide Correspondence to of Japanese, our study included 358 RA-ILD cases and association study (GWAS) in all populations. Dr Yukinori Okada, Department 4550 RA subjects without ILD. -

Annrheumdis-2019-216644.Full.Pdf
Crystal arthropathies Ann Rheum Dis: first published as 10.1136/annrheumdis-2019-216644 on 1 April 2020. Downloaded from EPIDEMIOLOGICAL SCIENCE Subtype- specific gout susceptibility loci and enrichment of selection pressure on ABCG2 and ALDH2 identified by subtype genome- wide meta- analyses of clinically defined gout patients Akiyoshi Nakayama,1,2 Masahiro Nakatochi ,3 Yusuke Kawamura,1,4 Ken Yamamoto,5 Hirofumi Nakaoka,6 Seiko Shimizu,1 Toshihide Higashino,1,7 Teruhide Koyama,8 Asahi Hishida,9 Kiyonori Kuriki,10 Miki Watanabe,11 Toru Shimizu,12,13 Keiko Ooyama,14 Hiroshi Ooyama,14 Mitsuo Nagase,15 Yuji Hidaka,16 Daisuke Matsui,8 Takashi Tamura,9 Takeshi Nishiyama,11 Chisato Shimanoe,17,18 Sakurako Katsuura- Kamano,19 Naoyuki Takashima,20,21 Yuya Shirai,22,23 Makoto Kawaguchi,1,24 Mikiya Takao,1,25 Ryo Sugiyama,1 Yuzo Takada,26 Takahiro Nakamura,27 Hiroshi Nakashima,28 Masashi Tsunoda,28 Inaho Danjoh,29 Atsushi Hozawa,30 Kazuyoshi Hosomichi,31 Yu Toyoda,32 Yu Kubota,32 Tappei Takada,32 Hiroshi Suzuki,32 Blanka Stiburkova,33,34 Tanya J. Major,35 Tony R. Merriman,35 Nagato Kuriyama,8 Haruo Mikami,36 Toshiro Takezaki,37 Keitaro Matsuo,38,39 Sadao Suzuki,11 Tatsuo Hosoya,40,41 Yoichiro Kamatani,42,43 Michiaki Kubo,44 Kimiyoshi Ichida,40,45 Kenji Wakai,9 Ituro Inoue,6 Yukinori Okada ,22,46 Nariyoshi Shinomiya,1 Hirotaka Matsuo,1 on behalf of Japan Gout Genomics Handling editor Josef S Consortium (Japan Gout) Smolen ► Additional material is ABSTRact Key messages published online only. To view Objectives Genome- wide meta- analyses of clinically please visit the journal online defined gout were performed to identify subtype- (http:// dx. -

Memoirs Volume63
ISSN 0369-0369 VOLUME 63,2006 MEMOIRS OF THE I nstitute of S cientific and I ndustrial Research OSAKA UNIVERSITY Contents Foreword············································································································· 1 Outline of ISIR 1. Research Activities ······························································································ 2 2. Education··············································································································· 14 3. International Exchange ························································································ 15 4. Concluding Remarks······························································································ 16 Activities of Divisions Quantum Engineering ······························································································ 19 Advanced Materials Science & Technology ···························································· 57 Organic Molecular Science ······················································································ 115 Intelligent Systems Science ····················································································· 157 Biological Science ································································································· 193 Quantum Beam Science & Technology ··································································· 218 Activities of Centers Nanoscience Nanotechnology Center ······································································ -

A4x66頁/PDF/約3300KB
2018 年度 研 究 年 報 (2018 年 4 月~2019 年 3 月) 東京大学 人工物工学研究センター 平成 31 年 5 月 〒277-8568 千葉県柏市柏の葉 5-1-5 Tel 04-7136-4240 Fax 04-7136-4242 http://www.race.u-tokyo.ac.jp/ 東京大学 人工物工学研究センター 2018 年度研究年報 目次 1. 人工物工学研究センター2018 年度の活動報告 センター長・教授 越塚 誠一 1 1.1 2018 年度の概要 --------------------------------------------------------------------------------------------------- 3 1.2 活動の概要 ---------------------------------------------------------------------------------------------------------- 3 1.3 人工物を創出するための理解Ⅰ ------------------------------------------------------------------------------ 5 2. 研究部門の概要と研究内容紹介(2019 年 3 月 31 日現在) 6 2.1 社会の中の人工物工学研究部門 教授 増田 昌敬-- 7 准教授 沖田 泰良 特任准教授 梁 云峰 教授 奥田 洋司(兼) 教授 藤田 豊久(兼) 教授 六川 修一(兼) 准教授 山田 知典(兼) 准教授 西野 成昭(兼) 講師 愛知 正温(兼) 2.2 人工物と人との相互作用研究部門 教授 太田 順 ----------- 22 教授 鈴木 克幸 准教授 原 辰徳 助教 白藤 翔平 教授 淺間 一(兼) 2.3 客員人工物工学研究部門 客員教授 中島 憲宏 ------- 41 客員准教授 青木 恵子 3. 講演会等の開催 ------------------------------------------------------------------------------------------------------------ 44 3.1 人工物工学研究センター主催の行事 -------------------------------------------------------------------- 45 3.1.1 RACE 研究発表会 ----------------------------------------------------------------------------------------- 45 3.2 人工物工学研究センター共同主催,共催の行事 ---------------------------------------------------- 46 3.2.1 構想設計革新イニシアティブ第Ⅱ回シンポジウム ------------------------------------------ 46 3.2.2 エッグドロップ甲子園 --------------------------------------------------------------------------------- 48 3.2.3 計算科学セミナー ------------------------------------------------------------------------------------- 50 4.