Discovery and Fine-Mapping of Kidney Function Loci in First Genome-Wide Association Study in Africans
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Homozygous Deletion of Six Olfactory Receptor Genes in a Subset of Individuals with Beta-Thalassemia
Homozygous Deletion of Six Olfactory Receptor Genes in a Subset of Individuals with Beta-Thalassemia Jessica Van Ziffle, Wendy Yang, Farid F. Chehab* Department of Laboratory Medicine, University of California San Francisco, San Francisco, California, United States of America Abstract Progress in the functional studies of human olfactory receptors has been largely hampered by the lack of a reliable experimental model system. Although transgenic approaches in mice could characterize the function of individual olfactory receptors, the presence of over 300 functional genes in the human genome becomes a daunting task. Thus, the characterization of individuals with a genetic susceptibility to altered olfaction coupled with the absence of particular olfactory receptor genes will allow phenotype/genotype correlations and vindicate the function of specific olfactory receptors with their cognate ligands. We characterized a 118 kb b-globin deletion and found that its 39 end breakpoint extends to the neighboring olfactory receptor region downstream of the b-globin gene cluster. This deletion encompasses six contiguous olfactory receptor genes (OR51V1, OR52Z1, OR51A1P, OR52A1, OR52A5, and OR52A4) all of which are expressed in the brain. Topology analysis of the encoded proteins from these olfactory receptor genes revealed that OR52Z1, OR52A1, OR52A5, and OR52A4 are predicted to be functional receptors as they display integral characteristics of G- proteins coupled receptors. Individuals homozygous for the 118 kb b-globin deletion are afflicted with b-thalassemia due to a homozygous deletion of the b-globin gene and have no alleles for the above mentioned olfactory receptors genes. This is the first example of a homozygous deletion of olfactory receptor genes in human. -

Cell Type Specificity of Chromatin Organization Mediated by CTCF
Cell type specificity of chromatin organization mediated by CTCF and cohesin Chunhui Hou, Ryan Dale, and Ann Dean1 Laboratory of Cellular and Developmental Biology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892 Edited* by Gary Felsenfeld, National Institutes of Health, Bethesda, MD, and approved January 7, 2010 (received for review October 20, 2009) CTCF sites are abundant in the genomes of diverse species but their be required for normal globin gene regulation in erythroid pro- function is enigmatic. We used chromosome conformation capture genitor cells (10). These functions of CTCF all involve CTCF- to determine long-range interactions among CTCF/cohesin sites mediated long-range chromatin organization, which can be man- over 2 Mb on human chromosome 11 encompassing the β-globin ifest intra- or interchromosomally (11, 12). locus and flanking olfactory receptor genes. Although CTCF occu- In a β-globin transgenic mouse model, we showed that CTCF- pies these sites in both erythroid K562 cells and fibroblast 293T cells, binding HS5 has intrinsic, portable enhancer blocking activity (13). the long-range interaction frequencies among the sites are highly When an extra copy of HS5 was placed between the locus control cell type specific, revealing a more densely clustered organization in region (LCR) and downstream genes, it formed a new insulator the absence of globin gene activity. Both CTCF and cohesins are loop with endogenous HS5 that topologically isolated the LCR required for the cell-type-specific chromatin conformation. Further- and nullified its activity. Here we show that CTCF/cohesin sites more, loss of the organizational loops in K562 cells through reduc- over a 2-Mb region of human chromosome 11, primarily among tion of CTCF with shRNA results in acquisition of repressive histone silent odorant receptor genes up- and downstream of the β-globin marks in the globin locus and reduces globin gene expression locus, participate in numerous interactions that form loops. -

Single Cell Derived Clonal Analysis of Human Glioblastoma Links
SUPPLEMENTARY INFORMATION: Single cell derived clonal analysis of human glioblastoma links functional and genomic heterogeneity ! Mona Meyer*, Jüri Reimand*, Xiaoyang Lan, Renee Head, Xueming Zhu, Michelle Kushida, Jane Bayani, Jessica C. Pressey, Anath Lionel, Ian D. Clarke, Michael Cusimano, Jeremy Squire, Stephen Scherer, Mark Bernstein, Melanie A. Woodin, Gary D. Bader**, and Peter B. Dirks**! ! * These authors contributed equally to this work.! ** Correspondence: [email protected] or [email protected]! ! Supplementary information - Meyer, Reimand et al. Supplementary methods" 4" Patient samples and fluorescence activated cell sorting (FACS)! 4! Differentiation! 4! Immunocytochemistry and EdU Imaging! 4! Proliferation! 5! Western blotting ! 5! Temozolomide treatment! 5! NCI drug library screen! 6! Orthotopic injections! 6! Immunohistochemistry on tumor sections! 6! Promoter methylation of MGMT! 6! Fluorescence in situ Hybridization (FISH)! 7! SNP6 microarray analysis and genome segmentation! 7! Calling copy number alterations! 8! Mapping altered genome segments to genes! 8! Recurrently altered genes with clonal variability! 9! Global analyses of copy number alterations! 9! Phylogenetic analysis of copy number alterations! 10! Microarray analysis! 10! Gene expression differences of TMZ resistant and sensitive clones of GBM-482! 10! Reverse transcription-PCR analyses! 11! Tumor subtype analysis of TMZ-sensitive and resistant clones! 11! Pathway analysis of gene expression in the TMZ-sensitive clone of GBM-482! 11! Supplementary figures and tables" 13" "2 Supplementary information - Meyer, Reimand et al. Table S1: Individual clones from all patient tumors are tumorigenic. ! 14! Fig. S1: clonal tumorigenicity.! 15! Fig. S2: clonal heterogeneity of EGFR and PTEN expression.! 20! Fig. S3: clonal heterogeneity of proliferation.! 21! Fig. -

Genetic Characterization of Greek Population Isolates Reveals Strong Genetic Drift at Missense and Trait-Associated Variants
ARTICLE Received 22 Apr 2014 | Accepted 22 Sep 2014 | Published 6 Nov 2014 DOI: 10.1038/ncomms6345 OPEN Genetic characterization of Greek population isolates reveals strong genetic drift at missense and trait-associated variants Kalliope Panoutsopoulou1,*, Konstantinos Hatzikotoulas1,*, Dionysia Kiara Xifara2,3, Vincenza Colonna4, Aliki-Eleni Farmaki5, Graham R.S. Ritchie1,6, Lorraine Southam1,2, Arthur Gilly1, Ioanna Tachmazidou1, Segun Fatumo1,7,8, Angela Matchan1, Nigel W. Rayner1,2,9, Ioanna Ntalla5,10, Massimo Mezzavilla1,11, Yuan Chen1, Chrysoula Kiagiadaki12, Eleni Zengini13,14, Vasiliki Mamakou13,15, Antonis Athanasiadis16, Margarita Giannakopoulou17, Vassiliki-Eirini Kariakli5, Rebecca N. Nsubuga18, Alex Karabarinde18, Manjinder Sandhu1,8, Gil McVean2, Chris Tyler-Smith1, Emmanouil Tsafantakis12, Maria Karaleftheri16, Yali Xue1, George Dedoussis5 & Eleftheria Zeggini1 Isolated populations are emerging as a powerful study design in the search for low-frequency and rare variant associations with complex phenotypes. Here we genotype 2,296 samples from two isolated Greek populations, the Pomak villages (HELIC-Pomak) in the North of Greece and the Mylopotamos villages (HELIC-MANOLIS) in Crete. We compare their genomic characteristics to the general Greek population and establish them as genetic isolates. In the MANOLIS cohort, we observe an enrichment of missense variants among the variants that have drifted up in frequency by more than fivefold. In the Pomak cohort, we find novel associations at variants on chr11p15.4 showing large allele frequency increases (from 0.2% in the general Greek population to 4.6% in the isolate) with haematological traits, for example, with mean corpuscular volume (rs7116019, P ¼ 2.3 Â 10 À 26). We replicate this association in a second set of Pomak samples (combined P ¼ 2.0 Â 10 À 36). -

Original Article a Pilot Study Comparing the Genetic Molecular Biology of Gestational and Non-Gestational Choriocarcinoma
Am J Transl Res 2019;11(11):7049-7062 www.ajtr.org /ISSN:1943-8141/AJTR0101016 Original Article A pilot study comparing the genetic molecular biology of gestational and non-gestational choriocarcinoma Cordelle Lazare1*, Wenhua Zhi1*, Jun Dai1, Canhui Cao1, Rajiv Rai Sookha1, Ling Wang1, Yifan Meng1, Peipei Gao1, Ping Wu1, Juncheng Wei1,2, Junbo Hu1, Peng Wu1,2 1The Key Laboratory of Cancer Invasion and Metastasis of The Ministry of Education of China, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, China; 2Department of Gynecologic Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, China. *Equal contributors and co-first authors. Received July 18, 2019; Accepted October 30, 2019; Epub November 15, 2019; Published November 30, 2019 Abstract: Non-gestational choriocarcinoma (NGC) is a rare subtype of choriocarcinoma differing in origin and phe- notypic characteristics compared to gestational choriocarcinoma (GC). This study aimed to analyze the molecular biology of GC and NGC and evaluate genetic anomalies of choriocarcinoma subtypes. DNA was extracted and paired from tumor-normal tissue of one NGC and one GC (control) patient for whole-exome sequencing. To further under- stand the role of DNAJB9, a p53 regulator mutated in the NGC tumor, on p53 upregulation in choriocarcinoma, CRISPR/Cas9 was used to induce DNAJB9 site-specific mutations in choriocarcinoma cells JEG-3. We hypothesized that DNAJB9 dysfunction would result in p53 overexpression. Sequencing revealed the GC tumor contained > 7 times more somatic mutations than the NGC tumor. Missense (98.86% vs. 94.97%), stop-gain (0.57% vs. -

Whole-Genome DNA Methylation and Hydroxymethylation Profiling for HBV-Related Hepatocellular Carcinoma
INTERNATIONAL JOURNAL OF ONCOLOGY 49: 589-602, 2016 Whole-genome DNA methylation and hydroxymethylation profiling for HBV-related hepatocellular carcinoma CHAO YE*, RAN TAO*, QINGYI CAO, DANHUA ZHU, YINI WANG, JIE WANG, JUAN LU, ERMEI CHEN and LANJUAN LI State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310000, P.R. China Received March 18, 2016; Accepted May 13, 2016 DOI: 10.3892/ijo.2016.3535 Abstract. Hepatocellular carcinoma (HCC) is a common tions between them. Taken together, in the present study we solid tumor worldwide with a poor prognosis. Accumulating conducted the first genome-wide mapping of DNA methyla- evidence has implicated important regulatory roles of epigen- tion combined with hydroxymethylation in HBV-related HCC etic modifications in the occurrence and progression of HCC. and provided a series of potential novel epigenetic biomarkers In the present study, we analyzed 5-methylcytosine (5-mC) for HCC. and 5-hydroxymethylcytosine (5-hmC) levels in the tumor tissues and paired adjacent peritumor tissues (APTs) from Introduction four individual HCC patients using a (hydroxy)methylated DNA immunoprecipitation approach combined with deep Hepatocellular carcinoma (HCC), a common solid tumor, is sequencing [(h)MeDIP-Seq]. Bioinformatics analysis revealed the third most frequent cause of cancer-related death in the that the 5-mC levels in the promoter regions of 2796 genes and world. Hepatitis B virus (HBV) infection is the main cause of the 5-hmC levels in 507 genes differed significantly between HCC in China (1). -

Supplementary Data
SUPPLEMENTARY METHODS 1) Characterisation of OCCC cell line gene expression profiles using Prediction Analysis for Microarrays (PAM) The ovarian cancer dataset from Hendrix et al (25) was used to predict the phenotypes of the cell lines used in this study. Hendrix et al (25) analysed a series of 103 ovarian samples using the Affymetrix U133A array platform (GEO: GSE6008). This dataset comprises clear cell (n=8), endometrioid (n=37), mucinous (n=13) and serous epithelial (n=41) primary ovarian carcinomas and samples from 4 normal ovaries. To build the predictor, the Prediction Analysis of Microarrays (PAM) package in R environment was employed (http://rss.acs.unt.edu/Rdoc/library/pamr/html/00Index.html). When more than one probe described the expression of a given gene, we used the probe with the highest median absolute deviation across the samples. The dataset from Hendrix et al. (25) and the dataset of OCCC cell lines described in this manuscript were then overlaid on the basis of 11536 common unique HGNC gene symbols. Only the 99 primary ovarian cancers samples and the four normal ovary samples were used to build the predictor. Following leave one out cross-validation, a predictor based upon 126 genes was able to identify correctly the four distinct phenotypes of primary ovarian tumour samples with a misclassification rate of 18.3%. This predictor was subsequently applied to the expression data from the 12 OCCC cell lines to determine the likeliest phenotype of the OCCC cell lines compared to primary ovarian cancers. Posterior probabilities were estimated for each cell line in comparison to the following phenotypes: clear cell, endometrioid, mucinous and serous epithelial. -

A Genome-Wide Association Study of Age-Related Hearing Impairment in Middle- and Old-Aged Chinese Twins
Hindawi BioMed Research International Volume 2021, Article ID 3629624, 14 pages https://doi.org/10.1155/2021/3629624 Research Article A Genome-Wide Association Study of Age-Related Hearing Impairment in Middle- and Old-Aged Chinese Twins Haiping Duan ,1,2,3 Wanxue Song ,1 Weijing Wang ,1 Hainan Cao ,4 Bingling Wang ,2,3 Yan Liu ,2,3 Chunsheng Xu ,2,3 Yili Wu ,1 Zengchang Pang ,2 and Dongfeng Zhang 1 1Department of Epidemiology and Health Statistics, Public Health College, Qingdao University, No. 38 Dengzhou Road, Shibei District, Qingdao, 266021 Shandong Province, China 2Qingdao Municipal Center for Disease Control and Prevention, No. 175 Shandong Road, Shibei District, Qingdao, 266033 Shandong Province, China 3Qingdao Institute of Preventive Medicine, No. 175 Shandong Road, Shibei District, Qingdao, 266033 Shandong Province, China 4Department of Otorhinolaryngology, Qingdao Municipal Hospital, Qingdao, 266011 Shandong Province, China Correspondence should be addressed to Dongfeng Zhang; [email protected] Received 23 July 2020; Revised 17 June 2021; Accepted 3 July 2021; Published 19 July 2021 Academic Editor: Cheol Lee Copyright © 2021 Haiping Duan et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Background. Age-related hearing impairment (ARHI) is considered an unpreventable disorder. We aimed to detect specific genetic variants that are potentially related to ARHI via genome-wide association study (GWAS). Methods. A sample of 131 dizygotic twins was genotyped for single-nucleotide polymorphism- (SNP-) based GWAS. Gene-based test was performed using VEGAS2. -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -

Supplementary Tables
Supplementary Tables Supplementary Table 1. Differentially methylated genes in correlation with their expression pattern in the A4 progression model A. Hypomethylated–upregulated Genes (n= 76) ALOX5 RRAD RTN4R DSCR6 FGFR3 HTR7 WNT3A POGK PLCD3 ALPPL2 RTEL1 SEMA3B DUSP5 FOSB ITGB4 MEST PPL PSMB8 ARHGEF4 BST2 SEMA7A SLC12A7 FOXQ1 KCTD12 LETM2 PRPH PXMP2 ARNTL2 CDH3 SHC2 SLC20A2 HSPA2 KIAA0182 LIMK2 NAB1 RASIP1 ASRGL1 CLDN3 DCBLD1 SNX10 SSH1 KREMEN2 LIPE NDRG2 ATF3 CLU DCHS1 SOD3 ST3GAL4 MAL LRRC1 NR3C2 ATP8B3 CYC1 DGCR8 EBAG9 SYNGR1 TYMS MCM2 NRG2 RHOF DAGLA DISP2 FAM19A5 TNNI3 UNC5B MYB PAK6 RIPK4 DAZAP1 DOCK3 FBXO6 HSPA4L WHSC1 PNMT PCDH1 B. Hypermethylated-downregulated Genes (n= 31) ARHGAP22 TNFSF9 KLF6 LRP8 NRP1 PAPSS2 SLC43A2 TBC1D16 ASB2 DZIP1 TPM1 MDGA1 NRP2 PIK3CD SMARCA2 TLL2 C18orf1 FBN1 LHFPL2 TRIO NTNG2 PTGIS SOCS2 TNFAIP8 DIXDC1 KIFC3 LMO1 NR3C1 ODZ3 PTPRM SYNPO Supplementary Table 2. Genes enriched for different histone methylation marks in A4 progression model identified through ChIP-on-chip a. H3K4me3 (n= 978) AATF C20orf149 CUL3 FOXP1 KATNA1 NEGR1 RAN SPIN2B ABCA7 C20orf52 CWF19L1 FRK KBTBD10 NEIL1 RANBP2 SPPL2A ABCC9 C21orf13-SH3BGR CXCL3 FSIP1 KBTBD6 NELF RAPGEF3 SPRY4 ABCG2 C21orf45 CYC1 FUK KCMF1 NFKB2 RARB SPRYD3 ABHD7 C22orf32 CYorf15A FXR2 KCNH7 NGDN RASAL2 SPTLC2 ACA15 C2orf18 DAXX FZD9 KCNMB4 NKAP RASD1 SRFBP1 ACA26 C2orf29 DAZ3 G6PD KCTD18 NKTR RASEF SRI ACA3 C2orf32 DBF4 GABPB2 KDELR2 NNT RASGRF1 SRM ACA48 C2orf55 DBF4B GABRA5 KIAA0100 NOL5A RASSF1 SSH2 ACAT1 C3orf44 DBI GADD45B KIAA0226 NOLC1 RASSF3 SSH3 ACSL5 -

Supplementary Materialsupplementary Material
DOI:10.1071/AN20275_AC CSIRO 2021 Animal Production Science 2021, 61, 731–744 Genome-wide association study between copy number variation regions and carcass and meat quality traits in Nellore cattle Mariana Piatto Berton1,5, Marcos Vinícius de Antunes Lemos1, Tatiane Cristina Seleguim Chud2, Nedenia Bonvino Stafuzza1, Sabrina Kluska1, Sabrina Thaise Amorim1, Lucas Silva Ferlin Lopes1, Angélica Simone Cravo Pereira3, Derek Bickhart4, George Liu4, Lúcia Galvão de Albuquerque1 and Fernando Baldi1 1Departamento de Zootecnia, Faculdade de Ciências Agrárias e Veterinárias, Universidade Estadual Paulista, Via de acesso Prof. Paulo Donato Castellane, s/no, CEP 14884-900 Jaboticabal, SP, Brazil 2Centre for Genetic Improvement of Livestock, University of Guelph, Guelph, ON, Canada N1G 2W1 3 Faculdade de Zootecnia e Engenharia de Alimentos, Universidade de São Paulo, Rua Duque de Caxias Norte, 225, CEP 13635-900 Pirassununga, SP, Brazil 4 USDA-ARS, ANRI, Bovine Functional Genomics Laboratory, Beltsville, Maryland 20705, USA; 2Department of Animal and Avian Sciences, University of Maryland, College Park, Maryland 20742, USA 5Corresponding author. E-mail: [email protected] Table S1. Description of significant (P value<0.05) CNVRs associated with beef tenderness (BT) and candidate genes within the CNVRs. CNVR ID BTA Type* Start (bp) End (bp) Size (bp) Low** High** P value FDR CNVR_gain_3 1 Gain 1374155 3404571 2030417 27 45 0.026 0.36 CNVR_gain_644 2 Gain 81291862 83154452 1862591 32 15 0.015 0.40 CNVR_loss_936 5 Loss 22514133 22563988 49856 233 -
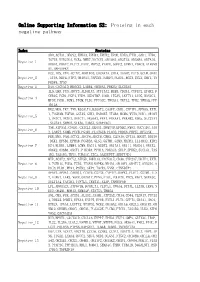
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2