RAPID-ACTING ANTIDEPRESSANTS, MARCH 2019 1 Rapid-Acting Antidepressants
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dextromethorphan Attenuates Sensorineural Hearing Loss in an Animal Model and Population-Based Cohort Study
International Journal of Environmental Research and Public Health Article Dextromethorphan Attenuates Sensorineural Hearing Loss in an Animal Model and Population-Based Cohort Study Hsin-Chien Chen 1,* , Chih-Hung Wang 1,2 , Wu-Chien Chien 3,4 , Chi-Hsiang Chung 3,4, Cheng-Ping Shih 1, Yi-Chun Lin 1,2, I-Hsun Li 5,6, Yuan-Yung Lin 1,2 and Chao-Yin Kuo 1 1 Department of Otolaryngology-Head and Neck Surgery, Tri-Service General Hospital, National Defense Medical Center, Taipei 114, Taiwan; [email protected] (C.-H.W.); [email protected] (C.-P.S.); [email protected] (Y.-C.L.); [email protected] (Y.-Y.L.); [email protected] (C.-Y.K.) 2 Graduate Institute of Medical Sciences, National Defense Medical Center, Taipei 114, Taiwan 3 School of Public Health, National Defense Medical Center, Taipei 114, Taiwan; [email protected] (W.-C.C.); [email protected] (C.-H.C.) 4 Department of Medical Research, Tri-Service General Hospital, National Defense Medical Center, Taipei 114, Taiwan 5 Department of Pharmacy Practice, Tri-Service General Hospital, National Defense Medical Center, Taipei 114, Taiwan; [email protected] 6 School of Pharmacy, National Defense Medical Center, Taipei 114, Taiwan * Correspondence: [email protected]; Tel.: +886-2-8792-7192; Fax: +886-2-8792-7193 Received: 31 July 2020; Accepted: 28 August 2020; Published: 31 August 2020 Abstract: The effect of dextromethorphan (DXM) use in sensorineural hearing loss (SNHL) has not been fully examined. We conducted an animal model and nationwide retrospective matched-cohort study to explore the association between DXM use and SNHL. -
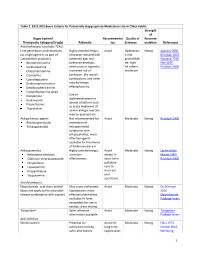
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

The Effect of Morphine-Lidocaine-Ketamine
THE EFFECT OF MORPHINE-LIDOCAINE-KETAMINE- DEXMEDETOMIDINE CO-INFUSION ON MINIMUM ALVEOLAR CONCENTRATION OF ISOFLURANE IN DOGS Thesis Presented in Partial Fulfillment of the Requirements for the Degree of Master of Science in the Graduate School of The Ohio State University By Lisa Michelle Sams, D.V.M. Graduate Program in Comparative and Veterinary Medicine The Ohio State University 2011 Thesis Committee: Dr. Phillip Lerche, Advisor Dr. Richard M. Bednarski Dr. John A.E. Hubbell Copyright by Lisa Michelle Sams 2011 ABSTRACT The purpose of this study was to determine the effects of an infusion of dexmedetomidine, a co-infusion of morphine-lidocaine-ketamine (MLK), and a co- infusion of dexmedetomidine-morphine-lidocaine-ketamine (alpha-MLK) on minimum alveolar concentration (MAC) of isoflurane in dogs. The MAC of an inhalant anesthetic required to prohibit purposeful movement is a measure of anesthetic potency (Eger et al 1965). Isoflurane is the most commonly used inhalant anesthetic in veterinary practice (Lozano et al 2009), but has potent vasodilatory effects and causes a dose-dependent decrease in mean arterial pressure in anesthetized dogs (Steffey and Howland 1977). Additional drugs are used during anesthesia to decrease the inhalant anesthetic requirement, a concept referred to as balanced anesthesia. Each of the drugs we infused has a different central nervous system receptor mechanism of action. Co-infusion of MLK, as well as the infusion of each drug separately, has been shown to reduce MAC in isoflurane-anesthetized dogs (Muir et al 2003). Dexmedetomidine, an alpha-2 agonist, has been shown to reduce isoflurane MAC in dogs (Pascoe et al 2006). -

RAP-MD-05 GLYX13-C-203 Protocol Amendment 3 Final-R
NCT02192099 Study ID: GLYX13‐C‐203 Title: Open Label Extension for Subjects with Inadequate/Partial Response to Antidepressants during the Current Episode of Major Depressive Disorder Previously Treated with Rapastinel (GLYX‐13) (Extension of GLYX13‐C‐202, NCT01684163) Protocol Amendment 3 Date: 23 Nov 2016 Clinical Study Protocol Protocol Number: GLYX13-C-203 Study Drug: Rapastinel (GLYX-13) Injection (rapastinel Injection) FDA ID: 107,974 Clinicaltrials.gov: Title: Open Label Extension for Subjects with Inadequate/Partial Response to Antidepressants during the Current Episode of Major Depressive Disorder Previously Treated with Rapastinel (GLYX-13) (Extension of GLYX13-C-202, NCT01684163) Study Phase: 2 Sponsor: Naurex, Inc, an affiliate of Allergan, plc. Date: Amendment 3 dated 23 November 2016 Replaces Amendment 2 dated 20 January 2015 This document is a confidential communication from Naurex, Inc, an affiliate of Allergan, plc.. Acceptance of this document constitutes an agreement by the recipient(s) that no information contained herein will be published or disclosed without prior written approval from Allergan, plc., except that this document may be disclosed to appropriate Institutional Review Boards (IRBs) under the condition that they are also required to maintain confidentiality. Naurex, Inc, an affiliate of Allergan, plc. Protocol GLYX13-C-203 INVESTIGATOR SIGNATURE PAGE The signature of the investigator below constitutes his/her approval of this protocol and provides the necessary assurances that this study will be conducted according to all stipulations of the protocol as specified in both the clinical and administrative sections, including all statements regarding confidentiality. This trial will be conducted in compliance with the protocol and all applicable regulatory requirements, in accordance with Good Clinical Practices (GCPs), including International Conference on Harmonization (ICH) Guidelines, and in general conformity with the most recent version of the Declaration of Helsinki. -

Antiemetics/Antivertigo Agents
Antiemetic Agents Therapeutic Class Review (TCR) May 1, 2019 No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage or retrieval system without the express written consent of Magellan Rx Management. All requests for permission should be mailed to: Magellan Rx Management Attention: Legal Department 6950 Columbia Gateway Drive Columbia, Maryland 21046 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Send comments and suggestions to [email protected]. May 2019 Proprietary Information. Restricted Access – Do not disseminate or copy without approval. © 2004-2019 Magellan Rx Management. All Rights Reserved. 3 FDA-APPROVED INDICATIONS Drug Manufacturer Indication(s) NK1 receptor antagonists aprepitant capsules generic, Merck In combination with other antiemetic agents for: (Emend®)1 . -

Scopolamine As a Potential Treatment Option in Major Depressive Disorder - a Literature Review
Isr J Psychiatry - Vol. 58 - No 1 (2021) Scopolamine as a Potential Treatment Option in Major Depressive Disorder - A Literature Review Dusan Kolar, MD, MSc, PhD, FRCPC Department of Psychiatry, Queen’s University, Mood Disorders Research and Treatment Service, Kingston, Ontario, Canada as selective serotonin reuptake inhibitors (SSRIs) and ABSTRACT serotonin norepinephrine reuptake inhibitors (SNRIs), are currently used as first-line pharmacological treat- Introduction: Slow onset response to antidepressants ments for major depressive disorder (MDD). Despite the and partial response is a common problem in mood availability of a comprehensive list of antidepressants, disorders psychiatry. There is an ongoing need for rapid- approximately 50% of the patients on an antidepressant acting antidepressants, particularly after introducing regimen experience non-response to treatment (3, 4). ketamine in the treatment of depression. Scopolamine Furthermore, the mood elevating effects of antidepres- may have promise as an antidepressant. sant medication is often delayed (5). It is recommended Methods: The author conducted a literature review to that the patient be treated for at least six weeks with identify available treatment trials of scopolamine in the adequate dosage of the given antidepressant before unipolar and bipolar depression in PubMed, the Cochrane considering making changes to the treatment (5). Partial database, Ovid Medline and Google Scholar. to no response to treatment with antidepressants and delayed onset of action indicate that there is a need for Results: There have been eight treatment trials of the development of novel and improved medications scopolamine in MDD and bipolar depression. Seven for the treatment of depression. Many studies in recent studies confirmed significant antidepressant effects years have recognised two different classes of drugs with of scopolamine used as monotherapy and as an rapid and robust antidepressant effects. -

The Promise of Ketamine
Neuropsychopharmacology (2015) 40, 257–258 & 2015 American College of Neuropsychopharmacology. All rights reserved 0893-133X/15 www.neuropsychopharmacology.org Editorial Circumspectives: The Promise of Ketamine William A Carlezon*,1 and Tony P George2 1 2 Department of Psychiatry, Harvard Medical School, McLean Hospital, Belmont, MA, USA; Department of Psychiatry, University of Toronto, Toronto, Ontario, Canada Neuropsychopharmacology (2015) 40, 257–258; doi:10.1038/npp.2014.270 In this issue we reprise a Feature called Circumspectives. offering hope that fast-acting but safe antidepressants are The general format of a Circumspectives article is similar to possible. a debate, with separate sections in which two thought Despite growing enthusiasm for ketamine and its leaders articulate their individual positions on a topic of promise, Dr Schatzberg describes some sobering details great importance to our community of researchers. The and gaps in knowledge. Ketamine is a drug of abuse and, distinguishing element, however, is that the piece ends with despite some exceptionally elegant studies on the mechan- a ‘reconciliation’ that is co-authored by both and includes ism (eg, Li et al, 2010; Autry et al, 2011), there is no ideas or experiments that will move the field forward. consensus on how it produces therapeutic effects. As one The current Circumspectives (Sanacora and Schatzberg, example, Dr Schatzberg points out similarities in some of 2015) is entitled ‘Ketamine: Promising Path or False the molecular actions of ketamine and scopolamine, Prophecy in the Development of Novel Therapeutics for another familiar and long-standing member of our phama- Mood Disorders?’. It is co-authored by Gerard Sanacora and copea shown to produce rapid antidepressant effects (Furey Alan F Schatzberg, who are leaders in this field. -

Poster Session III, December 6, 2017
Neuropsychopharmacology (2017) 42, S476–S652 © 2017 American College of Neuropsychopharmacology. All rights reserved 0893-133X/17 www.neuropsychopharmacology.org Poster Session III a high-resolution research tomograph (HRRT), and struc- Palm Springs, California, December 3–7, 2017 tural (T1) MRI using a 3-Tesla scanner. PET data analyses were carried out using the validated 2-tissue compartment Sponsorship Statement: Publication of this supplement is model to determine the total volume of distribution (VT) of sponsored by the ACNP. [18F]FEPPA. Individual contributor disclosures may be found within the Results: Results show significant inverse associations (con- abstracts. Part 1: All Financial Involvement with a pharma- trolling for rs6971 genotype) between neuroinflammation ceutical or biotechnology company, a company providing (VTs) and cortical thickness in the right medial prefrontal clinical assessment, scientific, or medical products or compa- cortex [r = -.562, p = .029] and the left dorsolateral nies doing business with or proposing to do business with prefrontal cortex [r = -.629, p = .012](p values are not ACNP over past 2 years (Calendar Years 2014–Present); Part 2: corrected for multiple comparisons). No significant associa- Income Sources & Equity of $10,000 per year or greater tions were found with surface area. (Calendar Years 2014 - Present): List those financial relation- Conclusions: These results, while preliminary, suggest links ships which are listed in part one and have a value greater than between the microglial activation/neuroinflammation and $10,000 per year, OR financial holdings that are listed in part cortical thickness in the dorsolateral prefrontal cortex and one and have a value of $10,000 or greater as of the date of medial prefrontal cortex in AD patients. -

Hallucinogens
Hallucinogens What are hallucinogens? Hallucinogens are a diverse group of drugs that alter a person’s awareness of their surroundings as well as their own thoughts and feelings. They are commonly split into two categories: classic hallucinogens (such as LSD) and dissociative drugs (such as PCP). Both types of hallucinogens can cause hallucinations, or sensations and images that seem real though they are not. Additionally, dissociative drugs can cause users to feel out of control or disconnected from their body and environment. Some hallucinogens are extracted from plants or mushrooms, and some are synthetic (human- made). Historically, people have used hallucinogens for religious or healing rituals. More recently, people report using these drugs for social or recreational purposes, including to have fun, deal with stress, have spiritual experiences, or just to feel different. Common classic hallucinogens include the following: • LSD (D-lysergic acid diethylamide) is one of the most powerful mind-altering chemicals. It is a clear or white odorless material made from lysergic acid, which is found in a fungus that grows on rye and other grains. LSD has many other street names, including acid, blotter acid, dots, and mellow yellow. • Psilocybin (4-phosphoryloxy-N,N- dimethyltryptamine) comes from certain types of mushrooms found in tropical and subtropical regions of South America, Mexico, and the United States. Some common names for Blotter sheet of LSD-soaked paper squares that users psilocybin include little smoke, magic put in their mouths. mushrooms, and shrooms. Photo by © DEA • Peyote (mescaline) is a small, spineless cactus with mescaline as its main ingredient. Peyote can also be synthetic. -
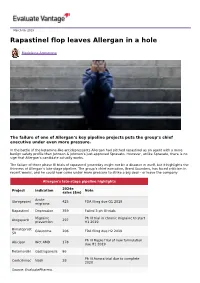
Rapastinel Flop Leaves Allergan in a Hole
March 06, 2019 Rapastinel flop leaves Allergan in a hole Madeleine Armstrong The failure of one of Allergan’s key pipeline projects puts the group’s chief executive under even more pressure. In the battle of the ketamine-like antidepressants Allergan had pitched rapastinel as an agent with a more benign safety profile than Johnson & Johnson’s just-approved Spravato. However, unlike Spravato, there is no sign that Allergan’s candidate actually works. The failure of three phase III trials of rapastinel yesterday might not be a disaster in itself, but it highlights the thinness of Allergan’s late-stage pipeline. The group’s chief executive, Brent Saunders, has faced criticism in recent weeks, and he could now come under more pressure to strike a big deal – or leave the company. Allergan's late-stage pipeline highlights 2024e Project Indication Note sales ($m) Acute Ubrogepant 425 FDA filing due Q1 2019 migraine Rapastinel Depression 359 Failed 3 ph III trials Migraine Ph III trial in chronic migraine to start Atogepant 297 prevention H1 2019 Bimatoprost Glaucoma 206 FDA filing due H2 2019 SR Ph III Maple trial of new formulation Abicipar Wet AMD 178 due H1 2019 Relamorelin Gastroparesis 86 Ph III Aurora trial due to complete Cenicriviroc Nash 28 2020 Source: EvaluatePharma. Activist shareholders have long been agitating for change at Allergan, and the latest failure will only provide more ammunition to those who want to split the roles of chairman and chief exec amid dissatisfaction with the group's acquisition strategy, and with its stock hovering around a five-year low. -

Antidepressant & Psychedelic Drug Interaction Chart
Copyright Psychedelic School 8/2020 Antidepressant & Psychedelic Drug Interaction Chart This chart is not intended to be used to make medical decisions and is for informational purposes only. It was constructed using data whenever possible, although extrapolation from known information was also used to inform risk. Any decision to start, stop, or taper medication and/or use psychedelic drugs should be made in conjunction with your healthcare provider(s). It is recommended to not perform any illicit activity. This chart the intellectual property of psychedelic school and is for personal use only. Please do not copy or distribute this chart. Antidepressant Phenethylamines Tryptamines MAOI-containing Ketamine Ibogaine -MDMA, mescaline -Psilocybin, LSD -Ayahuasca, Syrian Rue SSRIs Taper & discontinue at least 2 Consider taper & Taper & discontinue at least 2 Has been studied and Taper & discontinue at · Paroxetine (Paxil) weeks prior (all except discontinuation at least 2 weeks weeks prior (all except found effective both with least 2 weeks prior (all · Sertraline (Zoloft) fluoxetine) or 6 weeks prior prior (all except fluoxetine) or 6 fluoxetine) or 6 weeks prior and without concurrent except fluoxetine) or 6 · Citalopram (Celexa) (fluoxetine only) due to loss of weeks prior (fluoxetine only) (fluoxetine only) due to use of antidepressants weeks prior (fluoxetine · Escitalopram (Lexapro) psychedelic effect due to potential loss of potential risk of serotonin only) due to risk of · Fluxoetine (Prozac) psychedelic effect syndrome additive QTc interval · Fluvoxamine (Luvox) MDMA is unable to cause Recommended prolongation, release of serotonin when the Chronic antidepressant use may Life threatening toxicities can to be used in conjunction arrhythmias, or SPARI serotonin reuptake pump is result in down-regulation of occur with these with oral antidepressants cardiotoxicity · Vibryyd (Vilazodone) blocked. -

WO 2017/066590 Al 20 April 2017 (20.04.2017) P O P CT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/066590 Al 20 April 2017 (20.04.2017) P O P CT (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 38/06 (2006.01) A61K 31/519 (2006.01) kind of national protection available): AE, AG, AL, AM, A61 38/07 (2006.0V) A61K 31/551 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 31/13 (2006.01) A61K 31/5513 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, A61K 31/135 (2006.01) A61P 25/24 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, A61K 31/496 (2006.01) A61P 25/18 (2006.01) HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US20 16/057071 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, 14 October 2016 (14.10.201 6) TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, zw. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/242,633 16 October 2015 (16.