Conservation and Divergence of Bhlh Genes in the Calcisponge Sycon Ciliatum Sofia A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Spicule Formation in Calcareous Sponges: Coordinated Expression
www.nature.com/scientificreports OPEN Spicule formation in calcareous sponges: Coordinated expression of biomineralization genes and Received: 17 November 2016 Accepted: 02 March 2017 spicule-type specific genes Published: 13 April 2017 Oliver Voigt1, Maja Adamska2, Marcin Adamski2, André Kittelmann1, Lukardis Wencker1 & Gert Wörheide1,3,4 The ability to form mineral structures under biological control is widespread among animals. In several species, specific proteins have been shown to be involved in biomineralization, but it is uncertain how they influence the shape of the growing biomineral and the resulting skeleton. Calcareous sponges are the only sponges that form calcitic spicules, which, based on the number of rays (actines) are distinguished in diactines, triactines and tetractines. Each actine is formed by only two cells, called sclerocytes. Little is known about biomineralization proteins in calcareous sponges, other than that specific carbonic anhydrases (CAs) have been identified, and that uncharacterized Asx-rich proteins have been isolated from calcitic spicules. By RNA-Seq and RNA in situ hybridization (ISH), we identified five additional biomineralization genes inSycon ciliatum: two bicarbonate transporters (BCTs) and three Asx-rich extracellular matrix proteins (ARPs). We show that these biomineralization genes are expressed in a coordinated pattern during spicule formation. Furthermore, two of the ARPs are spicule- type specific for triactines and tetractines (ARP1 orSciTriactinin ) or diactines (ARP2 or SciDiactinin). Our results suggest that spicule formation is controlled by defined temporal and spatial expression of spicule-type specific sets of biomineralization genes. By the process of biomineralization many animal groups produce mineral structures like skeletons, shells and teeth. Biominerals differ in shape considerably from their inorganic mineral counterparts1. -

Review of the Mineralogy of Calcifying Sponges
Dickinson College Dickinson Scholar Faculty and Staff Publications By Year Faculty and Staff Publications 12-2013 Not All Sponges Will Thrive in a High-CO2 Ocean: Review of the Mineralogy of Calcifying Sponges Abigail M. Smith Jade Berman Marcus M. Key, Jr. Dickinson College David J. Winter Follow this and additional works at: https://scholar.dickinson.edu/faculty_publications Part of the Paleontology Commons Recommended Citation Smith, Abigail M.; Berman, Jade; Key,, Marcus M. Jr.; and Winter, David J., "Not All Sponges Will Thrive in a High-CO2 Ocean: Review of the Mineralogy of Calcifying Sponges" (2013). Dickinson College Faculty Publications. Paper 338. https://scholar.dickinson.edu/faculty_publications/338 This article is brought to you for free and open access by Dickinson Scholar. It has been accepted for inclusion by an authorized administrator. For more information, please contact [email protected]. © 2013. Licensed under the Creative Commons http://creativecommons.org/licenses/by- nc-nd/4.0/ Elsevier Editorial System(tm) for Palaeogeography, Palaeoclimatology, Palaeoecology Manuscript Draft Manuscript Number: PALAEO7348R1 Title: Not all sponges will thrive in a high-CO2 ocean: Review of the mineralogy of calcifying sponges Article Type: Research Paper Keywords: sponges; Porifera; ocean acidification; calcite; aragonite; skeletal biomineralogy Corresponding Author: Dr. Abigail M Smith, PhD Corresponding Author's Institution: University of Otago First Author: Abigail M Smith, PhD Order of Authors: Abigail M Smith, PhD; Jade Berman, PhD; Marcus M Key Jr, PhD; David J Winter, PhD Abstract: Most marine sponges precipitate silicate skeletal elements, and it has been predicted that they would be among the few "winners" in an acidifying, high-CO2 ocean. -

Modeling the Phenotype of Spinal Muscular Atrophy by the Direct Conversion of Human Fibroblasts to Motor Neurons
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 7), pp: 10945-10953 Research Paper Modeling the phenotype of spinal muscular atrophy by the direct conversion of human fibroblasts to motor neurons Qi-Jie Zhang1,*, Jin-Jing Li1,*, Xiang Lin1, Ying-Qian Lu1, Xin-Xin Guo1, En-Lin Dong1, Miao Zhao1, Jin He1, Ning Wang1,2 and Wan-Jin Chen1,2 1 Department of Neurology and Institute of Neurology, First Affiliated Hospital, Fujian Medical University, Fuzhou, China 2 Fujian Key Laboratory of Molecular Neurology, Fuzhou, China * These authors have contributed equally to this work Correspondence to: Wan-Jin Chen, email: [email protected] Keywords: direct reprogramming; fibroblast; induced motor neuron; spinal muscular atrophy Received: June 08, 2016 Accepted: November 22, 2016 Published: January 13, 2017 ABSTRACT Spinal muscular atrophy (SMA) is a lethal autosomal recessive neurological disease characterized by selective degeneration of motor neurons in the spinal cord. In recent years, the development of cellular reprogramming technology has provided an alternative and effective method for obtaining patient-specific neuronsin vitro. In the present study, we applied this technology to the field of SMA to acquire patient- specific induced motor neurons that were directly converted from fibroblasts via the forced expression of 8 defined transcription factors. The infected fibroblasts began to grow in a dipolar manner, and the nuclei gradually enlarged. Typical Tuj1-positive neurons were generated at day 23. After day 35, induced neurons with multiple neurites were observed, and these neurons also expressed the hallmarks of Tuj1, HB9, ISL1 and CHAT. The conversion efficiencies were approximately 5.8% and 5.5% in the SMA and control groups, respectively. -

Belgian Register of Marine Species
BELGIAN REGISTER OF MARINE SPECIES September 2010 Belgian Register of Marine Species – September 2010 BELGIAN REGISTER OF MARINE SPECIES, COMPILED AND VALIDATED BY THE VLIZ BELGIAN MARINE SPECIES CONSORTIUM VLIZ SPECIAL PUBLICATION 46 SUGGESTED CITATION Leen Vandepitte, Wim Decock & Jan Mees (eds) (2010). Belgian Register of Marine Species, compiled and validated by the VLIZ Belgian Marine Species Consortium. VLIZ Special Publication, 46. Vlaams Instituut voor de Zee (VLIZ): Oostende, Belgium. 78 pp. ISBN 978‐90‐812900‐8‐1. CONTACT INFORMATION Flanders Marine Institute – VLIZ InnovOcean site Wandelaarkaai 7 8400 Oostende Belgium Phone: ++32‐(0)59‐34 21 30 Fax: ++32‐(0)59‐34 21 31 E‐mail: [email protected] or [email protected] ‐ 2 ‐ Belgian Register of Marine Species – September 2010 Content Introduction ......................................................................................................................................... ‐ 5 ‐ Used terminology and definitions ....................................................................................................... ‐ 7 ‐ Belgian Register of Marine Species in numbers .................................................................................. ‐ 9 ‐ Belgian Register of Marine Species ................................................................................................... ‐ 12 ‐ BACTERIA ............................................................................................................................................. ‐ 12 ‐ PROTOZOA ........................................................................................................................................... -

Regulation of Adult Neurogenesis in Mammalian Brain
International Journal of Molecular Sciences Review Regulation of Adult Neurogenesis in Mammalian Brain 1,2, 3, 3,4 Maria Victoria Niklison-Chirou y, Massimiliano Agostini y, Ivano Amelio and Gerry Melino 3,* 1 Centre for Therapeutic Innovation (CTI-Bath), Department of Pharmacy & Pharmacology, University of Bath, Bath BA2 7AY, UK; [email protected] 2 Blizard Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, UK 3 Department of Experimental Medicine, TOR, University of Rome “Tor Vergata”, 00133 Rome, Italy; [email protected] (M.A.); [email protected] (I.A.) 4 School of Life Sciences, University of Nottingham, Nottingham NG7 2HU, UK * Correspondence: [email protected] These authors contributed equally to this work. y Received: 18 May 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: Adult neurogenesis is a multistage process by which neurons are generated and integrated into existing neuronal circuits. In the adult brain, neurogenesis is mainly localized in two specialized niches, the subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) adjacent to the lateral ventricles. Neurogenesis plays a fundamental role in postnatal brain, where it is required for neuronal plasticity. Moreover, perturbation of adult neurogenesis contributes to several human diseases, including cognitive impairment and neurodegenerative diseases. The interplay between extrinsic and intrinsic factors is fundamental in regulating neurogenesis. Over the past decades, several studies on intrinsic pathways, including transcription factors, have highlighted their fundamental role in regulating every stage of neurogenesis. However, it is likely that transcriptional regulation is part of a more sophisticated regulatory network, which includes epigenetic modifications, non-coding RNAs and metabolic pathways. -

E Proteins and ID Proteins: Helix-Loop-Helix Partners in Development and Disease
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Developmental Cell Review E Proteins and ID Proteins: Helix-Loop-Helix Partners in Development and Disease Lan-Hsin Wang1 and Nicholas E. Baker1,2,3,* 1Department of Genetics, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA 2Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA 3Department of Ophthalmology and Visual Sciences, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA *Correspondence: [email protected] http://dx.doi.org/10.1016/j.devcel.2015.10.019 The basic Helix-Loop-Helix (bHLH) proteins represent a well-known class of transcriptional regulators. Many bHLH proteins act as heterodimers with members of a class of ubiquitous partners, the E proteins. A widely expressed class of inhibitory heterodimer partners—the Inhibitor of DNA-binding (ID) proteins—also exists. Genetic and molecular analyses in humans and in knockout mice implicate E proteins and ID proteins in a wide variety of diseases, belying the notion that they are non-specific partner proteins. Here, we explore relationships of E proteins and ID proteins to a variety of disease processes and highlight gaps in knowledge of disease mechanisms. E proteins and Inhibitor of DNA-binding (ID) proteins are widely conferring DNA-binding specificity and transcriptional activation expressed transcriptional regulators with very general functions. on heterodimers with the ubiquitous E proteins (Figure 1). They are implicated in diseases by evidence ranging from Another class of pervasive HLH proteins acts in opposition to confirmed Mendelian inheritance, association studies, and E proteins. -

Lineage Transcription Factors Co-Regulate Subtype-Specific Genes Providing a Roadmap For
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.13.249029; this version posted August 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Lineage transcription factors co-regulate subtype-specific genes providing a roadmap for systematic identification of small cell lung cancer vulnerabilities Karine Pozo1,2, Rahul K. Kollipara3, Demetra P. Kelenis1, Kathia E. Rodarte1, Xiaoyang Zhang4, John D. Minna5,6,7,8 and Jane E. Johnson1,6,8 1Department of Neuroscience, 2Department of Surgery, 3McDermott Center for Human Growth and Development, UT Southwestern Medical Center, Dallas, TX 75390, USA 4Department of Oncological Sciences, Huntsman Cancer Institute, University of Utah, Salt Lake City, UT 5Hamon Center for Therapeutic Oncology Research, 6Simmons Comprehensive Cancer Center, 7Department of Internal Medicine, 8Department of Pharmacology, UT Southwestern Medical Center, Dallas, TX 75390, USA JDM receives licensing fees for lung cancer lines from the NIH and UT Southwestern. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.08.13.249029; this version posted August 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. ABSTRACT Lineage-defining transcription factors (LTFs) play key roles in tumor cell growth, making them highly attractive, but currently “undruggable”, small cell lung cancer (SCLC) vulnerabilities. -

Sponge Fauna in the Sea of Marmara
www.trjfas.org ISSN 1303-2712 Turkish Journal of Fisheries and Aquatic Sciences 16: 51-59 (2016) DOI: 10.4194/1303-2712-v16_1_06 RESEARCH PAPER Sponge Fauna in the Sea of Marmara Bülent Topaloğlu1,*, Alper Evcen2, Melih Ertan Çınar2 1 Istanbul University, Department of Marine Biology, Faculty of Fisheries, 34131 Vezneciler, İstanbul, Turkey. 2 Ege University, Department of Hydrobiology, Faculty of Fisheries, 35100 Bornova, İzmir, Turkey. * Corresponding Author: Tel.: +90.533 2157727; Fax: +90.512 40379; Received 04 December 2015 E-mail: [email protected] Accepted 08 February 2016 Abstract Sponge species collected along the coasts of the Sea of the Marmara in 2012-2013 were identified. A total of 30 species belonging to 21 families were found, of which four species (Ascandra contorta, Paraleucilla magna, Raspailia (Parasyringella) agnata and Polymastia penicillus) are new records for the eastern Mediterranean, while six species [A. contorta, P. magna, Chalinula renieroides, P. penicillus, R. (P.) agnata and Spongia (Spongia) nitens] are new records for the marine fauna of Turkey and 12 species are new records for the Sea of Marmara. Sponge specimens were generally collected in shallow water, but two species (Thenea muricata and Rhizaxinella elongata) were found at depths deeper than 100 m. One alien species (P. magna) was found at 10 m depth at station K18 (Büyükada). The morphological and distributional features of the species that are new to the Turkish marine fauna are presented. Keywords: Porifera, Benthos, Invertebrate, Turkish Straits System. Marmara Denizi Sünger Faunası Özet Bu çalışmada, Marmara Denizi ve kıyılarında 2012-2013 yılları arasında toplanan Sünger örnekleri tanımlanmıştır. -

Basal Metazoans - Dirk Erpenbeck, Simion Paul, Michael Manuel, Paulyn Cartwright, Oliver Voigt and Gert Worheide
EVOLUTION OF PHYLOGENETIC TREE OF LIFE - Basal Metazoans - Dirk Erpenbeck, Simion Paul, Michael Manuel, Paulyn Cartwright, Oliver Voigt and Gert Worheide BASAL METAZOANS Dirk Erpenbeck Ludwig-Maximilians Universität München, Germany Simion Paul and Michaël Manuel Université Pierre et Marie Curie in Paris, France. Paulyn Cartwright University of Kansas USA. Oliver Voigt and Gert Wörheide Ludwig-Maximilians Universität München, Germany Keywords: Metazoa, Porifera, sponges, Placozoa, Cnidaria, anthozoans, jellyfishes, Ctenophora, comb jellies Contents 1. Introduction on ―Basal Metazoans‖ 2. Phylogenetic relationships among non-bilaterian Metazoa 3. Porifera (Sponges) 4. Placozoa 5. Ctenophora (Comb-jellies) 6. Cnidaria 7. Cultural impact and relevance to human welfare Glossary Bibliography Biographical Sketch Summary Basal metazoans comprise the four non-bilaterian animal phyla Porifera (sponges), Cnidaria (anthozoans and jellyfishes), Placozoa (Trichoplax) and Ctenophora (comb jellies). The phylogenetic position of these taxa in the animal tree is pivotal for our understanding of the last common metazoan ancestor and the character evolution all Metazoa,UNESCO-EOLSS but is much debated. Morphological, evolutionary, internal and external phylogenetic aspects of the four phyla are highlighted and discussed. SAMPLE CHAPTERS 1. Introduction on “Basal Metazoans” In many textbooks the term ―lower metazoans‖ still refers to an undefined assemblage of invertebrate phyla, whose phylogenetic relationships were rather undefined. This assemblage may contain both bilaterian and non-bilaterian taxa. Currently, ―Basal Metazoa‖ refers to non-bilaterian animals only, four phyla that lack obvious bilateral symmetry, Porifera, Placozoa, Cnidaria and Ctenophora. ©Encyclopedia of Life Support Systems (EOLSS) EVOLUTION OF PHYLOGENETIC TREE OF LIFE - Basal Metazoans - Dirk Erpenbeck, Simion Paul, Michael Manuel, Paulyn Cartwright, Oliver Voigt and Gert Worheide These four phyla have classically been known as ―diploblastic‖ Metazoa. -

Retinoblastoma Tumor Suppressor Protein in Pancreatic Progenitors Controls Α- and Β-Cell Fate
Retinoblastoma tumor suppressor protein in pancreatic progenitors controls α- and β-cell fate Erica P. Caia,b, Xiaohong Wuc, Stephanie A. Schroera, Andrew J. Eliad,e, M. Cristina Nostroa,f, Eldad Zacksenhausa, and Minna Wooa,b,e,g,1 aToronto General Research Institute and fMcEwen Centre for Regenerative Medicine, University Health Network, Toronto, ON, Canada M5G 1L7; bInstitute of Medical Science, University of Toronto, Toronto, ON, Canada M5S 1A8; cThe First Affiliated Hospital, Nanjing Medical University, Nanjing, Jiangsu 210029, China; dCampbell Family Institute for Breast Cancer Research, University Health Network, Toronto, ON, Canada M5G 2M9; eDepartment of Medical Biophysics, University of Toronto, Toronto, ON, Canada M5G 2M9; and gDivision of Endocrinology, Department of Medicine, Toronto General Hospital, University Health Network, University of Toronto, Toronto, ON, Canada M5G 2C4 Edited by Tak W. Mak, The Campbell Family Institute for Breast Cancer Research, Ontario Cancer Institute at Princess Margaret Hospital, University Health Network, Toronto, ON, Canada, and approved July 16, 2013 (received for review February 20, 2013) Pancreatic endocrine cells expand rapidly during embryogenesis apoptosis (15), in a highly cell-specific and context-dependent by neogenesis and proliferation, but during adulthood, islet cells manner (16, 17). Although Rb is critical in regulating cell cycle have a very slow turnover. Disruption of murine retinoblastoma entry in proliferating cells, its role in postmitotic cells is more tumor suppressor protein (Rb) in mature pancreatic β-cells has limited. For example, Rb deficiency in proliferating myoblasts a limited effect on cell proliferation. Here we show that deletion induces increased proliferation and apoptosis (18), whereas fi fi of Rb during embryogenesis in islet progenitors leads to an in- Rb de ciency in postmitotic muscle bers does not lead to any crease in the neurogenin 3-expressing precursor cell population, defects (19). -
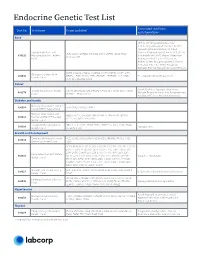
Endocrine Genetic Test List
Endocrine Genetic Test List Associated conditions Test No. Test Name Genes Included and phenotypes Bone HPP, X-Linked Hypophosphatemia, X-Linked Hypophosphatemic Rickets, XLH, Hypophosphatemic Rickets, X-Linked Hypophosphatasia and Dominant Hypophosphatemic Rickets (XLHR), ALPL, CLCN5, CYP2R1, CYP27B1, DMP1, ENPP1, FGF23, PHEX, 630292 Hypophosphatemic Rickets X-Linked Rickets (XLR), Vitamin D-Resistant SLC34A3, VDR Panel Rickets, X-Linked Vitamin D-Resistant Rickets (VDRR), Hypophosphatemic Vitamin D-Resistant Rickets (HPDR), Phosphate Diabetes, Familial Hypophosphatemic Rickets BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LRP5, Osteogenesis Imperfecta 630543 MBTPS2, P3H1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, SPARC, OI, Juvenile Primary Osteoporosis Genetic Panel TENT5A, TMEM38B, WNT1 Cancer Familial Isolated Hyperparathyroidism, VistaSeq® Endocrine Cancer CDC73, MAX, MEN1, NF1, PRKAR1A, PTEN, RET, SDHB, SDHC, SDHD, 481374 Multiple Endocrine Neoplasia, Paraganglioma, Panel* TMEM127, TP53 and VHL Parathyroid Cancer, Pheochromocytoma Diabetes and Insulin Maturity-Onset Diabetes of the 630568 GCK, HNF1A, HNF1B, HNF4A Young (MODY) 4-gene Panel Maturity-Onset Diabetes of ABCC8, APPL1, BLK, GCK, HNF1A, HNF1B, HNF4A, INS, KCNJ11, 630513 the Young (MODY) Expanded KLF11, NEUROD1, PAX4, PDX1 Genetic Panel Congenital Hyperinsulinism ABCC8, GCK, GLUD1, HADH, HNF1A, HNF4A, KCNJ11, PGM1, PMM2, 630500 Hypoglycemia Genetic Panel SLC16A1, UCP2 Growth and Development Combined Pituitary Hormone GLI1, HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, -

The Evolution of the Mitochondrial Proteome in Animals
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2019 The evolution of the mitochondrial proteome in animals Viraj Muthye Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Bioinformatics Commons Recommended Citation Muthye, Viraj, "The evolution of the mitochondrial proteome in animals" (2019). Graduate Theses and Dissertations. 17752. https://lib.dr.iastate.edu/etd/17752 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. The evolution of the mitochondrial proteome in animals by Viraj Rajendra Muthye A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Bioinformatics and Computational Biology Program of Study Committee: Dennis Lavrov, Co-major Professor Carolyn Lawrence-Dill, Co-major Professor Karin Dorman Robert Jernigan Iddo Friedberg The student author, whose presentation of the scholarship herein was approved by the program of study committee, is solely responsible for the content of this dissertation. The Graduate College will ensure this dissertation is globally accessible and will not permit alterations after a degree is conferred. Iowa State University Ames, Iowa 2019 Copyright c Viraj Rajendra Muthye, 2019. All rights reserved. ii DEDICATION To my wife, younger brother, parents and friends for their unconditional support, commitment and encouragement throughout my life iii TABLE OF CONTENTS Page ACKNOWLEDGMENTS .