List Item Oncaspar : EPAR
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Infection of the CNS by Scedosporium Apiospermum After Near Drowning
205 CASE REPORT J Clin Pathol: first published as 10.1136/jcp.2003.8680 on 27 January 2004. Downloaded from Infection of the CNS by Scedosporium apiospermum after near drowning. Report of a fatal case and analysis of its confounding factors P A Kowacs, C E Soares Silvado, S Monteiro de Almeida, M Ramos, K Abra˜o, L E Madaloso, R L Pinheiro, L C Werneck ............................................................................................................................... J Clin Pathol 2004;57:205–207. doi: 10.1136/jcp.2003.8680 from the usual 15 days to up to 130 days. This type of This report describes a fatal case of central nervous system infection causes granulomata or abscesses and neutrophilic pseudallescheriasis. A 32 year old white man presented with meningitis.125 headache and meningismus 15 days after nearly drowning in a swine sewage reservoir. Computerised tomography and ‘‘In cases secondary to aspiration after near drowning, magnetic resonance imaging of the head revealed multiple once in the bloodstream, fungi seed into several sites but brain granulomata, which vanished when steroid and broad develop mainly in the central nervous system’’ spectrum antimicrobial and antifungal agents, in addition to dexamethasone, were started. Cerebrospinal fluid analysis To date, few cases of CNS pseudallescheriasis have been 2 disclosed a neutrophilic meningitis. Treatment with antibiotics described. However, such a diagnosis must should always be sought in individuals who have suffered near drowning in and amphotericin B, together with fluconazole and later standing polluted streams, ponds of water or sewage, or pits itraconazole, was ineffective. Miconazole was added with manure. through an Ommaya reservoir, but was insufficient to halt The case of a man who acquired a CNS P boydii infection the infection. -

BC Cancer Benefit Drug List September 2021
Page 1 of 65 BC Cancer Benefit Drug List September 2021 DEFINITIONS Class I Reimbursed for active cancer or approved treatment or approved indication only. Reimbursed for approved indications only. Completion of the BC Cancer Compassionate Access Program Application (formerly Undesignated Indication Form) is necessary to Restricted Funding (R) provide the appropriate clinical information for each patient. NOTES 1. BC Cancer will reimburse, to the Communities Oncology Network hospital pharmacy, the actual acquisition cost of a Benefit Drug, up to the maximum price as determined by BC Cancer, based on the current brand and contract price. Please contact the OSCAR Hotline at 1-888-355-0355 if more information is required. 2. Not Otherwise Specified (NOS) code only applicable to Class I drugs where indicated. 3. Intrahepatic use of chemotherapy drugs is not reimbursable unless specified. 4. For queries regarding other indications not specified, please contact the BC Cancer Compassionate Access Program Office at 604.877.6000 x 6277 or [email protected] DOSAGE TUMOUR PROTOCOL DRUG APPROVED INDICATIONS CLASS NOTES FORM SITE CODES Therapy for Metastatic Castration-Sensitive Prostate Cancer using abiraterone tablet Genitourinary UGUMCSPABI* R Abiraterone and Prednisone Palliative Therapy for Metastatic Castration Resistant Prostate Cancer abiraterone tablet Genitourinary UGUPABI R Using Abiraterone and prednisone acitretin capsule Lymphoma reversal of early dysplastic and neoplastic stem changes LYNOS I first-line treatment of epidermal -

Australian Public Assessment Report for Pegaspargase
Australian Public Assessment Report for pegaspargase Proprietary Product Name: Oncaspar Sponsor: Baxalta Australia September 2018 Therapeutic Goods Administration About the Therapeutic Goods Administration (TGA) • The Therapeutic Goods Administration (TGA) is part of the Australian Government Department of Health and is responsible for regulating medicines and medical devices. • The TGA administers the Therapeutic Goods Act 1989 (the Act), applying a risk management approach designed to ensure therapeutic goods supplied in Australia meet acceptable standards of quality, safety and efficacy (performance) when necessary. • The work of the TGA is based on applying scientific and clinical expertise to decision- making, to ensure that the benefits to consumers outweigh any risks associated with the use of medicines and medical devices. • The TGA relies on the public, healthcare professionals and industry to report problems with medicines or medical devices. TGA investigates reports received by it to determine any necessary regulatory action. • To report a problem with a medicine or medical device, please see the information on the TGA website < https://www.tga.gov.au>. About AusPARs • An Australian Public Assessment Report (AusPAR) provides information about the evaluation of a prescription medicine and the considerations that led the TGA to approve or not approve a prescription medicine submission. • AusPARs are prepared and published by the TGA. • An AusPAR is prepared for submissions that relate to new chemical entities, generic medicines, major variations and extensions of indications. • An AusPAR is a static document; it provides information that relates to a submission at a particular point in time. • A new AusPAR will be developed to reflect changes to indications and/or major variations to a prescription medicine subject to evaluation by the TGA. -

Adverse Events After Immunisation- Common and Uncommon
Adverse events following Immunisation Common and Uncommon Dr Anna Clarke National Immunisation Office September 2016 www.immunisation.ie Abbreviations • ADR-adverse drug reaction • AE- adverse event • AEFI-adverse event following immunisation • SAE- serious adverse event • SUSAR-suspected unexpected serious adverse reaction Definitions • Adverse Drug Reaction – A response to a drug which is noxious and unintended, …occurs at doses normally used for the prophylaxis,.. or therapy of disease, … • Adverse Event – Any untoward medical occurrence that may present during treatment with a pharmaceutical product but which does not necessarily have a causal relationship with this treatment Definitions Adverse Event Following Immunization (AEFI) Any untoward medical occurrence which follows immunisation and which does not necessarily have a causal relationship with the usage of the vaccine. The adverse event may be any unfavourable or unintended sign, abnormal laboratory finding, symptom or disease. AEFI 1. Loose definition to encourage reporting - does not restrict type of event - does not limit the time after immunisation - events, not reactions, are reported 2. Belief that immunisation was responsible may be correct, incorrect, or impossible to assess 3. Does not imply causality AEFIs Mild Reactions – Common – Include pain, swelling, fever, irritability, malaise – Self-limiting, seldom requiring symptomatic treatment But - important to inform parents about such events so they know about them Serious Adverse Event • Fatal • Life-threatening • -
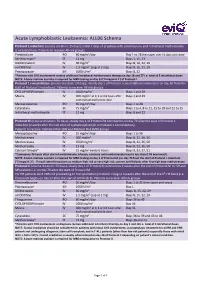
Acute Lymphoblastic Leukaemia: ALL06 Schema
Acute Lymphoblastic Leukaemia: ALL06 Schema Protocol 1 induction: (course duration: 35 days); initial 7 days of prephase with prednisolone and intrathecal methotrexate is included here. Patients to receive: All risk groups Prednisolone PO 60 mg/m2/day Day 1 to 28 then taper over 10 days and cease Methotrexate* IT 12 mg Days 1, 15, 33 DAUNOrubicin IV 30 mg/m2 Days 8, 16, 22, 29 vinCRISTine IV 1.5 mg/m2 (cap at 2 mg) Days 8, 16, 22, 29 Pegaspargase IM 1000 U/m2 Days 8, 22 *Patients with CNS involvement receive additional intrathecal methotrexate therapy on day 18 and 27 i.e. total of 5 intrathecal doses NOTE: A bone marrow aspirate is required for MRD testing on day 33 ('Timepoint 1') of Protocol I Protocol 1 consolidation: (course duration: 29 days); ideally day 1 of Protocol I consolidation commences on day 36 from the start of Protocol 1 induction ). Patients to receive: All risk groups. CYCLOPHOSPHamide IV 1000 mg/m2 Days 1 and 29 Mesna IV 400 mg/m2 at 0, 4 and 8 hours after Days 1 and 29 each cyclophosphamide dose Mercaptopurine PO 60 mg/m2/day Days 1 to 28 Cytarabine SC 75 mg/m2 Days 1 to 4, 8 to 11, 15 to 18 and 22 to 25 Intrathecal methotrexate IT 12 mg Days 8 and 22 Protocol M: (course duration: 56 days); ideally day 1 of Protocol M commences on day 79 after the start of Protocol 1 induction (2 weeks after the last dose of cyclophosphamide in Protocol 1 consolidation). -

Oncaspar, INN-Pegaspargase
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Oncaspar 750 U/ml solution for injection/infusion. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION One ml of solution contains 750 Units (U)** of pegaspargase*. One vial of 5 ml solution contains 3,750 Units. * The active substance is a covalent conjugate of Escherichia coli-derived L-asparaginase with monomethoxypolyethylene glycol **One unit is defined as the quantity of enzyme required to liberate 1 µmol ammonia per minute at pH 7.3 and 37°C The potency of this medicinal product should not be compared to the one of another pegylated or non-pegylated protein of the same therapeutic class. For more information, see section 5.1. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Solution for injection/infusion. Clear, colourless solution. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Oncaspar is indicated as a component of antineoplastic combination therapy in acute lymphoblastic leukaemia (ALL) in paediatric patients from birth to 18 years, and adult patients. 4.2 Posology and method of administration Oncaspar should be prescribed and administered by physicians and/or health care personnel experienced in the use of antineoplastic products. It should only be given in a hospital setting where appropriate resuscitation equipment is available. Patients should be closely monitored for any adverse reactions throughout the administration period (see section 4.4). Posology Oncaspar is usually administered as part of combination chemotherapy protocols with other antineoplastic agents (see also section 4.5). Paediatric patients and adults ≤21 years The recommended dose in patients with a body surface area (BSA) ≥0.6 m2 and who are ≤21 years of age is 2,500 U of pegaspargase (equivalent to 3.3 ml Oncaspar)/m2 body surface area every 14 days. -

Prescribing Information | VELCADE® (Bortezomib)
HIGHLIGHTS OF PRESCRIBING INFORMATION Hypotension: Use caution when treating patients taking These highlights do not include all the information needed to antihypertensives, with a history of syncope, or with dehydration. use VELCADE safely and effectively. See full prescribing (5.2) information for VELCADE. Cardiac Toxicity: Worsening of and development of cardiac VELCADE® (bortezomib) for injection, for subcutaneous or failure has occurred. Closely monitor patients with existing heart intravenous use disease or risk factors for heart disease. (5.3) Initial U.S. Approval: 2003 Pulmonary Toxicity: Acute respiratory syndromes have ------------------------RECENT MAJOR CHANGES-------------------------- occurred. Monitor closely for new or worsening symptoms and consider interrupting VELCADE therapy. (5.4) Warnings and Precautions, Posterior Reversible Encephalopathy Syndrome: Consider MRI Thrombotic Microangiopathy (5.10) 04/2019 imaging for onset of visual or neurological symptoms; ------------------------INDICATIONS AND USAGE-------------------------- discontinue VELCADE if suspected. (5.5) VELCADE is a proteasome inhibitor indicated for: Gastrointestinal Toxicity: Nausea, diarrhea, constipation, and treatment of adult patients with multiple myeloma (1.1) vomiting may require use of antiemetic and antidiarrheal medications or fluid replacement. (5.6) treatment of adult patients with mantle cell lymphoma (1.2) Thrombocytopenia and Neutropenia: Monitor complete blood ----------------------DOSAGE AND ADMINISTRATION--------------------- -

Reviewer Guidance
Reviewer Guidance Conducting a Clinical Safety Review of a New Product Application and Preparing a Report on the Review U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) February 2005 Good Review Practices Reviewer Guidance Conducting a Clinical Safety Review of a New Product Application and Preparing a Report on the Review Additional copies are available from: Office of Training and Communication Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 (Tel) 301-827-4573 http://www.fda.gov/cder/guidance/index.htm U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) February 2005 Good Review Practices TABLE OF CONTENTS I. INTRODUCTION............................................................................................................. 1 II. GENERAL GUIDANCE ON THE CLINICAL SAFETY REVIEW .......................... 2 A. Introduction....................................................................................................................................2 B. Explanation of Terms ....................................................................................................................3 C. Overview of the Safety Review .....................................................................................................4 D. Differences in Approach to Safety and Effectiveness Data ........................................................4 -
Acute Lymphoblastic Leukemia (ALL) (Part 1 Of
LEUKEMIA TREATMENT REGIMENS: Acute Lymphoblastic Leukemia (ALL) (Part 1 of 12) Note: The National Comprehensive Cancer Network (NCCN) Guidelines® for Acute Lymphoblastic Leukemia (ALL) should be consulted for the management of patients with lymphoblastic lymphoma. Clinical Trials: The NCCN recommends cancer patient participation in clinical trials as the gold standard for treatment. Cancer therapy selection, dosing, administration, and the management of related adverse events can be a complex process that should be handled by an experienced healthcare team. Clinicians must choose and verify treatment options based on the individual patient; drug dose modifications and supportive care interventions should be administered accordingly. The cancer treatment regimens below may include both U.S. Food and Drug Administration-approved and unapproved indications/regimens. These regimens are only provided to supplement the latest treatment strategies. The NCCN Guidelines are a work in progress that may be refined as often as new significant data becomes available. They are a consensus statement of its authors regarding their views of currently accepted approaches to treatment. Any clinician seeking to apply or consult any NCCN Guidelines is expected to use independent medical judgment in the context of individual clinical circumstances to determine any patient’s care or treatment. The NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any -

Negative Change Add Inclusion Criteria
Policy Drug(s) Type of Change Brief Description of Policy Change new policy Elitek (rasburicase) n/a n/a new policy Retevmo (selpercatinib) n/a n/a new policy Tabrecta (capmatinib) n/a n/a new policy Trodelvy (govitecan-hziy) n/a n/a new policy Qinlock (ripretinib) n/a n/a UM ONC_1048 Campath (alemtuzumab) Archive Campath is no longer being used for any Hematology Oncology indications. Remove inclusion criteria: ALL: Induction, consolidation, maitenance, relapsed/refractory combination removed and use is supported as a part of a multi-agent chemotherapy regimen, for all sub-types of ALL, for induction/consolidation therapy, and for therapy of relapsed refractory disease NHL: Induction, consolidation, maitenance, relapsed/refractory combination removed and use is supported for extra- nodal NK/T-cell lymphoma (nasal type) and as a part of a multi-agent chemotherapy regimen for either first line therapy and/or therapy for relapsed /refractory disease. UM ONC_1063 Oncaspar (pegaspargase) Positive change Remove exclusion criteria: History of serious thrombosis, pancreatitis, or hemorrhagic events with L-asparaginase (ELSPAR) therapy. UM ONC_1064 Oncaspar (pegaspargase) Positive change Add exclusion criteria: Dosing exceeds single dose limit of Trastuzumab 8 mg/kg for the loading dose, 6 mg/kg for UM ONC_1134 Trastuzumab products Negative change subsequent doses when given every 3 weeks; 4 mg/kg for the loading dose and 2 mg/kg for the weekly dose. Remove exclusion criteria: Velcade (bortezomib) is being used after disease progression on another Velcade-based regimen. UM ONC_1136 Velcade (bortezomib) Positive change Add inclusion criteria: Multiple Myeloma: - NOTE: The preferred agent, per NCH policy, is generic bortezomib over brand bortezomib (Velcade) for all indications, unless there are contraindications or intolerance to generic bortezomib. -

AHRQ Quality Indicators Fact Sheet
AHRQ Quality Indicators Toolkit Fact Sheet on Inpatient Quality Indicators What are the Inpatient Quality Indicators? The Inpatient Quality Indicators (IQIs) include 28 provider-level indicators established by the Agency for Healthcare Research and Quality (AHRQ) that can be used with hospital inpatient discharge data to provide a perspective on quality. They are grouped into the following four sets: • Volume indicators are proxy, or indirect, measures of quality based on counts of admissions during which certain intensive, high-technology, or highly complex procedures were performed. They are based on evidence suggesting that hospitals performing more of these procedures may have better outcomes. • Mortality indicators for inpatient procedures include procedures for which mortality has been shown to vary across institutions and for which there is evidence that high mortality may be associated with poorer quality of care. • Mortality indicators for inpatient conditions include conditions for which mortality has been shown to vary substantially across institutions and for which evidence suggests that high mortality may be associated with deficiencies in the quality of care. • Utilization indicators examine procedures whose use varies significantly across hospitals and for which questions have been raised about overuse, underuse, or misuse. Mortality for Selected Procedures and Mortality for Selected Conditions are composite measures that AHRQ established in 2008. Each composite is estimated as a weighted average, across a set of IQIs, of the ratio of a hospital’s observed rate (OR) to its expected rate (ER), based on a reference population: OR/ER. The IQI-specific ratios are adjusted for reliability before they are averaged, to minimize the influence of ratios that are high or low at a specific hospital by chance. -

Interventions for Treating Acute High Altitude Illness
This document is downloaded from DR‑NTU (https://dr.ntu.edu.sg) Nanyang Technological University, Singapore. Interventions for treating acute high altitude illness Simancas‑Racines, Daniel; Arevalo‑Rodriguez, Ingrid; Osorio, Dimelza; Franco, Juan V.A.; Xu, Yihan; Hidalgo, Ricardo 2018 Simancas‑Racines, D., Arevalo‑Rodriguez, I., Osorio, D., Franco, J. V., Xu, Y., & Hidalgo, R. Interventions for treating acute high altitude illness. Cochrane Database of Systematic Reviews. (6), CD009567‑. doi:10.1002/14651858.CD009567.pub2 https://hdl.handle.net/10356/82999 https://doi.org/10.1002/14651858.CD009567.pub2 © 2018 The Cochrane Collaboration. All rights reserved. This paper was published by John Wiley & Sons, Ltd. in Cochrane Database of Systematic Reviews and is made available with permission of The Cochrane Collaboration. Downloaded on 28 Sep 2021 20:37:23 SGT [Intervention Review] Interventions for treating acute high altitude illness Daniel Simancas-Racines1, Ingrid Arevalo-Rodriguez1,2,3, Dimelza Osorio1, Juan VA Franco4, Yihan Xu5, Ricardo Hidalgo1 1Cochrane Ecuador. Centro de Investigación en Salud Pública y Epidemiología Clínica (CISPEC). Facultad de Ciencias de la Salud Eugenio Espejo, Universidad Tecnológica Equinoccial, Quito, Ecuador. 2Clinical Biostatistics Unit, Hospital Ramon y Cajal (IRYCIS), Madrid, Spain. 3CIBER Epidemiología y Salud Pública (CIBERESP), Madrid, Spain. 4Argentine Cochrane Centre, Instituto Universi- tario Hospital Italiano, Buenos Aires, Argentina. 5Wee Kim Wee School of Communication and Information, Nanyang Technological University, Singapore City, Singapore Contact address: Daniel Simancas-Racines, Cochrane Ecuador. Centro de Investigación en Salud Pública y Epidemiología Clínica (CISPEC). Facultad de Ciencias de la Salud Eugenio Espejo, Universidad Tecnológica Equinoccial, Quito, Ecuador. [email protected], [email protected].