Gene Expression Profiling in the Leukemic Stem Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tandem Genomic Arrangement of a G Protein (Gna15) and G Protein-Coupled Receptor (S1p4/Lpc1/Edg6) Gene James J.A
FEBS 26595 FEBS Letters 531 (2002) 99^102 Tandem genomic arrangement of a G protein (Gna15) and G protein-coupled receptor (s1p4/lpC1/Edg6) gene James J.A. Contos, Xiaoqin Ye, Valerie P. Sah, Jerold Chunà Department of Pharmacology, School of Medicine, University of California at San Diego, La Jolla, CA 92093-0636, USA Received 31 May 2002; revised 30 July 2002; accepted 6 September 2002 First published online 18 September 2002 Edited by Edward A. Dennis, Isabel Varela-Nieto and Alicia Alonso Genomic structure analysis of s1p4 could provide insight Abstract A genomic analysis of the s1p4/lpC1/Edg6 mouse sphingosine-1-phosphate (S1P) G protein-coupled receptor into the evolution of the eight lysophospholipid receptor gene revealed it to be located on central chromosome 10 and genes. The coding regions for each of the lpa genes are divided to consist of two exons with an intronless coding region. Sur- between two exons, whereas for the s1p1À3 genes, the coding prisingly, we found the gene encoding the promiscuously cou- region of each gene is within single exon, with only non-cod- pling GK15 protein (Gna15) located in tandem just upstream, an ing exon(s) upstream [8^12]. One would expect the genomic arrangement conserved in the human genome (on chromosome structure of s1p4 to be similar to the other s1p genes. How- 19p13.3). Given that Northern blots demonstrated similar tissue ever, to date, genomic structure information has not been distributions of the mouse s1p and Gna15 transcripts, we pro- 4 reported for s1p4. pose that transcription of the two genes may be under control of Lysophospholipid receptors, as well as all GPCRs, couple the same enhancer elements and that their protein products may to heterotrimeric G proteins, which consist of K, L and Q sub- couple in vivo. -

The Human Genome Project
TO KNOW OURSELVES ❖ THE U.S. DEPARTMENT OF ENERGY AND THE HUMAN GENOME PROJECT JULY 1996 TO KNOW OURSELVES ❖ THE U.S. DEPARTMENT OF ENERGY AND THE HUMAN GENOME PROJECT JULY 1996 Contents FOREWORD . 2 THE GENOME PROJECT—WHY THE DOE? . 4 A bold but logical step INTRODUCING THE HUMAN GENOME . 6 The recipe for life Some definitions . 6 A plan of action . 8 EXPLORING THE GENOMIC LANDSCAPE . 10 Mapping the terrain Two giant steps: Chromosomes 16 and 19 . 12 Getting down to details: Sequencing the genome . 16 Shotguns and transposons . 20 How good is good enough? . 26 Sidebar: Tools of the Trade . 17 Sidebar: The Mighty Mouse . 24 BEYOND BIOLOGY . 27 Instrumentation and informatics Smaller is better—And other developments . 27 Dealing with the data . 30 ETHICAL, LEGAL, AND SOCIAL IMPLICATIONS . 32 An essential dimension of genome research Foreword T THE END OF THE ROAD in Little has been rapid, and it is now generally agreed Cottonwood Canyon, near Salt that this international project will produce Lake City, Alta is a place of the complete sequence of the human genome near-mythic renown among by the year 2005. A skiers. In time it may well And what is more important, the value assume similar status among molecular of the project also appears beyond doubt. geneticists. In December 1984, a conference Genome research is revolutionizing biology there, co-sponsored by the U.S. Department and biotechnology, and providing a vital of Energy, pondered a single question: Does thrust to the increasingly broad scope of the modern DNA research offer a way of detect- biological sciences. -

Genetic Associations with Clozapine-Induced Myocarditis in Patients with Schizophrenia Paul Lacaze 1, Kathlyn J
Lacaze et al. Translational Psychiatry (2020) 10:37 https://doi.org/10.1038/s41398-020-0722-0 Translational Psychiatry ARTICLE Open Access Genetic associations with clozapine-induced myocarditis in patients with schizophrenia Paul Lacaze 1, Kathlyn J. Ronaldson 1,EuniceJ.Zhang2,AnaAlfirevic2,HardikShah3,LeahNewman3, Maya Strahl3, Melissa Smith3,ChadBousman4,BenFrancis5, Andrew P. Morris5,6, Trevor Wilson7, Fernando Rossello8, David Powell9, Vivien Vasic7, Robert Sebra3,JohnJ.McNeil1 and Munir Pirmohamed2 Abstract Clozapine is the most effective antipsychotic drug for schizophrenia, yet it can cause life-threatening adverse drug reactions, including myocarditis. The aim of this study was to determine whether schizophrenia patients with clozapine-induced myocarditis have a genetic predisposition compared with clozapine-tolerant controls. We measured different types of genetic variation, including genome-wide single-nucleotide polymorphisms (SNPs), coding variants that alter protein expression, and variable forms of human leucocyte antigen (HLA) genes, alongside traditional clinical risk factors in 42 cases and 67 controls. We calculated a polygenic risk score (PRS) based on variation at 96 different genetic sites, to estimate the genetic liability to clozapine-induced myocarditis. Our genome-wide association analysis identified four SNPs suggestive of increased myocarditis risk (P <1×10−6), with odds ratios ranging 5.5–13.7. The SNP with the lowest P value was rs74675399 (chr19p13.3, P = 1.21 × 10−7;OR= 6.36), located in the GNA15 gene, previously associated with heart failure. The HLA-C*07:01 allele was identified as potentially predisposing to clozapine-induced myocarditis (OR = 2.89, 95% CI: 1.11–7.53), consistent with a previous report of association of the same allele with clozapine-induced agranulocytosis. -

RNA Sequencing of Mesenchymal Stem Cells Reveals a Blocking Of
Lamas et al. Arthritis Research & Therapy (2019) 21:112 https://doi.org/10.1186/s13075-019-1894-y RESEARCH ARTICLE Open Access RNA sequencing of mesenchymal stem cells reveals a blocking of differentiation and immunomodulatory activities under inflammatory conditions in rheumatoid arthritis patients Jose Ramon Lamas1, Benjamin Fernandez-Gutierrez1* , Arkaitz Mucientes1, Fernando Marco2, Yaiza Lopiz2, Juan Angel Jover1, Lydia Abasolo1 and Luis Rodríguez-Rodríguez1 Abstract Introduction: Mesenchymal stem cells (MSCs) have the ability to differentiate into different types of cells of the mesenchymal lineage, such as osteocytes, chondrocytes, and adipocytes. It is also known that under inflammatory stimuli or in the appropriate experimental conditions, they can also act as regulators of inflammation. Thus, in addition to their regenerating potential, their interest has been extended to their possible use in cell therapy strategies for treatment of immune disorders. Objective: To analyze, by RNA-seq analysis, the transcriptome profiling of allogenic MSCs under RA lymphocyte activation. Methods: We identified the differentially expressed genes in bone marrow mesenchymal stem cells after exposure to an inflammatory environment. The transcriptome profiling was evaluated by means of the precise measurement of transcripts provided by the RNA-Seq technology. Results: Our results evidenced the existence of blocking of both regenerative (differentiation) and immunomodulatory phenotypes under inflammatory conditions characterized by an upregulation of genes involved in immune processes and a simultaneous downregulation of genes mainly involved in regenerative or cell differentiation functions. Conclusions: We conclude that the two main functions of MSCs (immunomodulation and differentiation) are blocked, at least while the inflammation is being resolved. Inflammation, at least partially mediated by gamma-interferon, drives MSCs to a cellular distress adopting a defensive state. -

Long-Read Assembly of a Great Dane Genome Highlights the Contribution of GC-Rich Sequence 2 and Mobile Elements to Canine Genomes 3 4 Julia V
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.31.231761; this version posted July 31, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Long-read assembly of a Great Dane genome highlights the contribution of GC-rich sequence 2 and mobile elements to canine genomes 3 4 Julia V. Halo1,2*, Amanda L. Pendleton2*, Feichen Shen2*, Aurélien J. Doucet2, Thomas Derrien3, 5 Christophe Hitte3, Laura E. Kirby2, Bridget Myers2, Elzbieta Sliwerska2, Sarah Emery2, John V. 6 Moran2,4, Adam R. Boyko5, Jeffrey M. Kidd2,6,7 7 8 1Department of Biological Sciences, Bowling Green State University, Bowling Green, OH, USA 9 2Department of Human Genetics, University of Michigan, Ann Arbor, MI, USA 10 3Univ. Rennes 1, CNRS, IGDR – UMR 6290, F-35000 Rennes, France 11 4Department of Internal Medicine, University of Michigan, Ann Arbor, MI, USA 12 5Department of Biomedical Sciences, Cornell University, Ithaca, New York, USA. 13 6Department Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, 14 MI USA 15 16 *These authors contributed equally 17 18 7Correspondence should be addressed to Jeffrey M. Kidd at [email protected] 19 20 21 Key words 22 Canis familiaris, long-read assembly, mobile elements, structural variation 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.31.231761; this version posted July 31, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

An Integrative Genomic Analysis of the Longshanks Selection Experiment for Longer Limbs in Mice
bioRxiv preprint doi: https://doi.org/10.1101/378711; this version posted August 19, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Title: 2 An integrative genomic analysis of the Longshanks selection experiment for longer limbs in mice 3 Short Title: 4 Genomic response to selection for longer limbs 5 One-sentence summary: 6 Genome sequencing of mice selected for longer limbs reveals that rapid selection response is 7 due to both discrete loci and polygenic adaptation 8 Authors: 9 João P. L. Castro 1,*, Michelle N. Yancoskie 1,*, Marta Marchini 2, Stefanie Belohlavy 3, Marek 10 Kučka 1, William H. Beluch 1, Ronald Naumann 4, Isabella Skuplik 2, John Cobb 2, Nick H. 11 Barton 3, Campbell Rolian2,†, Yingguang Frank Chan 1,† 12 Affiliations: 13 1. Friedrich Miescher Laboratory of the Max Planck Society, Tübingen, Germany 14 2. University of Calgary, Calgary AB, Canada 15 3. IST Austria, Klosterneuburg, Austria 16 4. Max Planck Institute for Cell Biology and Genetics, Dresden, Germany 17 Corresponding author: 18 Campbell Rolian 19 Yingguang Frank Chan 20 * indicates equal contribution 21 † indicates equal contribution 22 Abstract: 23 Evolutionary studies are often limited by missing data that are critical to understanding the 24 history of selection. Selection experiments, which reproduce rapid evolution under controlled 25 conditions, are excellent tools to study how genomes evolve under strong selection. Here we 1 bioRxiv preprint doi: https://doi.org/10.1101/378711; this version posted August 19, 2018. -
Transcriptome-Wide Analysis of Differentially Expressed Chemokine Receptors, Snps, and Ssrs in the Age-Related Macular Degenerat
Saddala et al. Human Genomics (2019) 13:15 https://doi.org/10.1186/s40246-019-0199-1 PRIMARYRESEARCH Open Access Transcriptome-wide analysis of differentially expressed chemokine receptors, SNPs, and SSRs in the age-related macular degeneration Madhu Sudhana Saddala1,2†, Anton Lennikov1,2† , Anthony Mukwaya3, Lijuan Fan1,2, Zhengmao Hu4 and Hu Huang1,2,5* Abstract Background: Age-related macular degeneration (AMD) is the most common, progressive, and polygenic cause of irreversible visual impairment in the world. The molecular pathogenesis of the primary events of AMD is poorly understood. We have investigated a transcriptome-wide analysis of differential gene expression, single-nucleotide polymorphisms (SNPs), indels, and simple sequence repeats (SSRs) in datasets of the human peripheral retina and RPE-choroid-sclera control and AMD. Methods and results: Adaptors and unbiased components were removed and checked to ensure the quality of the data sets. Molecular function, biological process, cellular component, and pathway analyses were performed on differentially expressed genes. Analysis of the gene expression datasets identified 5011 upregulated genes, 11,800 downregulated genes, 42,016 SNPs, 1141 indels, and 6668 SRRs between healthy controls and AMD donor material. Enrichment categories for gene ontology included chemokine activity, cytokine activity, cytokine receptor binding, immune system process, and signal transduction respectively. A functional pathways analysis identified that chemokine receptors bind chemokines, complement cascade genes, and create cytokine signaling in immune system pathway genes (p value < 0.001). Finally, allele-specific expression was found to be significant for Chemokine (C-C motif) ligand (CCL) 2, 3, 4, 13, 19, 21; C-C chemokine receptor (CCR) 1, 5; chemokine (C-X-C motif) ligand (CXCL) 9, 10, 16; C-X-C chemokine receptor type (CXCR) 6; as well as atypical chemokine receptor (ACKR) 3,4 and pro-platelet basic protein (PPBP). -

Network Mining Approach to Cancer Biomarker Discovery
NETWORK MINING APPROACH TO CANCER BIOMARKER DISCOVERY THESIS Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Praneeth Uppalapati, B.E. Graduate Program in Computer Science and Engineering The Ohio State University 2010 Thesis Committee: Dr. Kun Huang, Advisor Dr. Raghu Machiraju Copyright by Praneeth Uppalapati 2010 ABSTRACT With the rapid development of high throughput gene expression profiling technology, molecule profiling has become a powerful tool to characterize disease subtypes and discover gene signatures. Most existing gene signature discovery methods apply statistical methods to select genes whose expression values can differentiate different subject groups. However, a drawback of these approaches is that the selected genes are not functionally related and hence cannot reveal biological mechanism behind the difference in the patient groups. Gene co-expression network analysis can be used to mine functionally related sets of genes that can be marked as potential biomarkers through survival analysis. We present an efficient heuristic algorithm EigenCut that exploits the properties of gene co- expression networks to mine functionally related and dense modules of genes. We apply this method to brain tumor (Glioblastoma Multiforme) study to obtain functionally related clusters. If functional groups of genes with predictive power on patient prognosis can be identified, insights on the mechanisms related to metastasis in GBM can be obtained and better therapeutical plan can be developed. We predicted potential biomarkers by dividing the patients into two groups based on their expression profiles over the genes in the clusters and comparing their survival outcome through survival analysis. -

Genetic Associations with Clozapine-Induced Myocarditis in Patients with Schizophrenia Paul Lacaze 1, Kathlyn J
Lacaze et al. Translational Psychiatry (2020) 10:37 https://doi.org/10.1038/s41398-020-0722-0 Translational Psychiatry ARTICLE Open Access Genetic associations with clozapine-induced myocarditis in patients with schizophrenia Paul Lacaze 1, Kathlyn J. Ronaldson 1,EuniceJ.Zhang2,AnaAlfirevic2,HardikShah3,LeahNewman3, Maya Strahl3, Melissa Smith3,ChadBousman4,BenFrancis5, Andrew P. Morris5,6, Trevor Wilson7, Fernando Rossello8, David Powell9, Vivien Vasic7, Robert Sebra3,JohnJ.McNeil1 and Munir Pirmohamed2 Abstract Clozapine is the most effective antipsychotic drug for schizophrenia, yet it can cause life-threatening adverse drug reactions, including myocarditis. The aim of this study was to determine whether schizophrenia patients with clozapine-induced myocarditis have a genetic predisposition compared with clozapine-tolerant controls. We measured different types of genetic variation, including genome-wide single-nucleotide polymorphisms (SNPs), coding variants that alter protein expression, and variable forms of human leucocyte antigen (HLA) genes, alongside traditional clinical risk factors in 42 cases and 67 controls. We calculated a polygenic risk score (PRS) based on variation at 96 different genetic sites, to estimate the genetic liability to clozapine-induced myocarditis. Our genome-wide association analysis identified four SNPs suggestive of increased myocarditis risk (P <1×10−6), with odds ratios ranging 5.5–13.7. The SNP with the lowest P value was rs74675399 (chr19p13.3, P = 1.21 × 10−7;OR= 6.36), located in the GNA15 gene, previously associated with heart failure. The HLA-C*07:01 allele was identified as potentially predisposing to clozapine-induced myocarditis (OR = 2.89, 95% CI: 1.11–7.53), consistent with a previous report of association of the same allele with clozapine-induced agranulocytosis. -

Genetic Analysis of Association Between Calcium Signaling and Hippocampal Activation, Memory Performance in the Young and Old, and Risk for Sporadic Alzheimer Disease
Supplementary Online Content Heck A, Fastenrath M, Coynel D, et al. Genetic analysis of association between calcium signaling and hippocampal activation, memory performance in the young and old, and risk for sporadic Alzheimer disease. JAMA Psychiatry. Published online September 2, 2015. doi:10.1001/jamapsychiatry.2015.1309. eAppendix. eFigure 1. Quantile-quantile plot for the Discovery sample. eFigure 2. Quantile-quantile plot for the Replication sample. eFigure 3. Quantile-quantile plot for the Zurich sample. eFigure 4. Quantile-quantile plot for AgeCoDe sample. eFigure 5. Brain activity related to EM independently of allelic load. eTable 1. Demographics of the cognitively healthy samples. eTable 2. Multilocus genetic score: Gene symbols, SNP IDs, reference allele, major allele and direction of effect on EM performance in the discovery sample. eTable 3. EM core gene set. eTable 4. Detailed information about the GSEA results in the discovery and the replication sample: gene P value, chromosome, gene start, gene end, most significant SNP, SNP P value, effect size, frequency. eReferences. This supplementary material has been provided by the authors to give readers additional information about their work. © 2015 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/02/2021 eAppendix. Detailed description of testing procedure in the Discovery sample: Subjects were presented 24 neutral, 24 positive and 24 aversive photographs in a random order. The photographs were taken from the international affective picture system (IAPS)1 and from an in-house database for eight of the neutral pictures that allowed us to equate the pictures for visual complexity and content (e.g., human presence). -
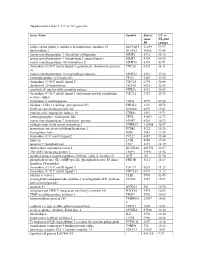
Supplemental Table 2: UC Vs. NL Gene List Gene Name Symbol
Supplemental Table 2: UC vs. NL gene list Gene Name Symbol Entrez UC vs. Gene NL fold ID change solute carrier family 6 (amino acid transporter), member 14 SLC6A14 11254 79.97 dual oxidase 2 DUOX2 50506 73.48 matrix metallopeptidase 1 (interstitial collagenase) MMP1 4312 65.16 matrix metallopeptidase 3 (stromelysin 1, progelatinase) MMP3 4314 60.20 matrix metallopeptidase 10 (stromelysin 2) MMP10 4319 40.71 chemokine (C-X-C motif) ligand 6 (granulocyte chemotactic protein CXCL6 6372 36.11 2) matrix metallopeptidase 12 (macrophage elastase) MMP12 4321 35.94 carboxypeptidase A3 (mast cell) CPA3 1359 33.94 chemokine (C-X-C motif) ligand 5 CXCL5 6374 30.84 cholesterol 25-hydroxylase CH25H 9023 28.08 myeloid cell nuclear differentiation antigen MNDA 4332 24.66 chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating CXCL1 2919 22.55 activity, alpha) tryptophan 2,3-dioxygenase TDO2 6999 21.66 chitinase 3-like 1 (cartilage glycoprotein-39) CHI3L1 1116 20.78 S100 calcium binding protein A8 S100A8 6279 19.81 cysteine-rich, angiogenic inducer, 61 CYR61 3491 19.71 carboxypeptidase, vitellogenic-like CPVL 54504 18.77 matrix metallopeptidase 7 (matrilysin, uterine) MMP7 4316 16.92 collagen triple helix repeat containing 1 CTHRC1 115908 15.47 proprotein convertase subtilisin/kexin type 1 PCSK1 5122 15.26 hemoglobin, beta HBB 3043 13.55 chemokine (C-C motif) ligand 2 CCL2 6347 13.44 lumican LUM 4060 13.43 tenascin C (hexabrachion) TNC 3371 12.19 dual oxidase maturation factor 2 DUOXA2 405753 12.07 TNFAIP3 interacting protein 3 TNIP3 79931 11.96