The Chemistry of Transactinide Elements — Experimental Achievements and Perspectives
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tables of Parameters Tables of Parameters for Extended Hückel Calculations Calculations
TABLES OF PARAMETERS FOR EXTENDED HÜCKEL CALCULATIONS Santiago Alvarez Universitat de Barcelona 1999 - 1 - PART 1. STANDARD Hii's and SLATER EXPONENTS AND COEFFICIENTS. H He Li Be B C N O F Ne Na Mg Al Si P S Cl Ar K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe Cs Ba Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn Fr Ra Rf Db Sg Bh Hs Mt La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr Parameters for all elements except the shaded ones are included in these Tables. - 2 - General Observations References are to the original paper which first reported EH calculations with the given Hii for a related set of molecules or solids. For some elements, when no EH calculations have been reported, the experimental ionization potentials are given and the references correspond to those experimental values. Although these tables are intended to be of some use in locating atomic parameters, reference to the original work is encouraged. Ionization potentials ( Hii) a) All energies are in eV. Hii's for most transition metals are charge-iterated for model compounds like metal carbonyls or typical organometallic molecules with Cp, CH3, CO, etc. Exceptions, including bulk metals are conveniently indicated. For cases in which charge-iterated Hii's are not available, the standard sources are the Hinze and Jaffé's tables of ionization potentials.1 b) When alternative sets of Hii's were available for the same element, the one which fits better with the trend along a group or a period of the periodic table has been chosen. -
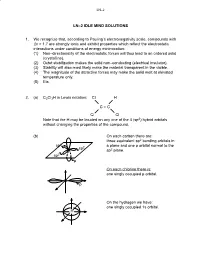
LN–2 IDLE MIND SOLUTIONS 1. We Recognize That, According To
LN–2 LN–2 IDLE MIND SOLUTIONS 1. We recognize that, according to Pauling’s electronegativity scale, compounds with Δx > 1.7 are strongly ionic and exhibit properties which reflect the electrostatic interactions under conditions of energy minimization: (1) Non–directionality of the electrostatic forces will thus lead to an ordered solid (crystalline). (2) Octet stabilization makes the solid non–conducting (electrical insulator). (3) Stability will also most likely make the material transparent in the visible. (4) The magnitude of the attractive forces may make the solid melt at elevated temperature only. (5) Etc. 2. (a) C2Cl3H in Lewis notation: Cl H C = C Cl Cl Note that the H may be located on any one of the 4 (sp2) hybrid orbitals without changing the properties of the compound. (b) On each carbon there are: three equivalent sp2 bonding orbitals in sp2 a plane and one p orbital normal to the sp2 sp2 plane. sp2 p On each chlorine there is: one singly occupied p orbital. Cl p On the hydrogen we have: one singly occupied 1s orbital. H LN–2 σ 2. (c) 2 sp2–sp2 orbitals → 1σ C–C π 2 p–p orbitals → 1π C–C 6 bonds 3 sp2–p orbitals → 3σ C–Cl 1 sp2–s orbital → 1σ C–H 3. Silicon (Si) is a group 4 element like C (carbon) and wil also exhibit sp3 hybridization: 4 sp3 orbitals at B.A. = 109o. Unlike carbon, silicon cannot form double or triple bonds (by lateral p orbital overlap) because the atom is too large to permit lateral overlap! (a) Si + 2Cl2 → SiCl4 Si is in the center of a tetrahedron with the chlorine atoms as the four corners. -

WO 2017/161171 A2 21 September 2017 (21.09.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/161171 A2 21 September 2017 (21.09.2017) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every B01J 23/10 (2006.01) B82Y 30/00 (201 1.01) kind of national protection available): AE, AG, AL, AM, B01J 35/02 (2006.01) C07C 2/84 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, (21) Number: International Application DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/US20 17/022787 HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KH, KN, (22) International Filing Date: KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, 16 March 2017 (16.03.2017) MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, (25) Filing Language: English RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, (26) Publication Language: English TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 62/309,284 16 March 2016 (16.03.2016) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: SILURIA TECHNOLOGIES, INC. GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, [US/US]; 409 Illinois Street, Suite 5032, San Francisco, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, California 94158 (US). -

Syntheses, Structures, Optical Properties, and Theoretical Calculations of Cs2bi2zns5,Cs2bi2cds5, and Cs2bi2mns5 Fu Qiang Huang, Rebecca C
ARTICLE IN PRESS Journal of Solid State Chemistry 174 (2003) 334–341 Syntheses, structures, optical properties, and theoretical calculations of Cs2Bi2ZnS5,Cs2Bi2CdS5, and Cs2Bi2MnS5 Fu Qiang Huang, Rebecca C. Somers, Adam D. McFarland, Richard P. Van Duyne, and James A Ibersà Department of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208-3113, USA Received 24 December 2002; received in revised form 14 April 2003; accepted 25 April 2003 Abstract Three new compounds, Cs2Bi2ZnS5,Cs2Bi2CdS5, and Cs2Bi2MnS5, have been synthesized from the respective elements and a reactive flux Cs2S3 at 973 K. The compounds are isostructural and crystallize in a new structure type in space group Pnma of the orthorhombic system with four formula units in cells of dimensions at 153 K of a=15.763(3), b=4.0965(9), c=18.197(4) A˚ , ˚ 3 ˚ ˚ 3 V=1175.0(4) A for Cs2Bi2ZnS5; a=15.817(2), b=4.1782(6), c=18.473(3) A, V=1220.8(3) A for Cs2Bi2CdS5; and a=15.830(2), ˚ ˚ 3 2 2À b=4.1515(5), c=18.372(2) A, V=1207.4(2) A for Cs2Bi2MnS5. The structure is composed of two-dimensional N[Bi2MS5 ] (M=Zn, Cd, Mn) layers that stack perpendicular to the [100] axis and are separated by Cs+ cations. The layers consist of edge- 1 6À 1 4À sharing N[Bi2S6 ] and N[MS3 ] chains built from BiS6 octahedral and MS4 tetrahedral units. Two crystallographically unique Cs atoms are coordinated to S atoms in octahedral and monocapped trigonal prismatic environments. The structure of Cs2Bi2MS5,is 0 0 related to that of Na2ZrCu2S4 and those of the AMM Q3 materials (A=alkali metal, M=rare-earth or Group 4 element, M = Group 11 or 12 element, Q=chalcogen). -

Macrocycles Containing 1, 1'-Ferrocenyldiselenolato Ligands On
Dalton Transactions View Article Online PAPER View Journal | View Issue Macrocycles containing 1,1’-ferrocenyldiselenolato ligands Cite this: Dalton Trans., 2018, 47, 5415 on group 4 metallocenes† Clara Sanchez-Perez,a,b Caroline E. Knapp, b Minna M. Karjalainen,a Raija Oilunkaniemi,a Claire J. Carmalt *b and Risto S. Laitinen *a 5 5 t Macrocyclic [Fe(η -C5H4Se)2M(η -C5H4R)2]2 [M = Ti (1), Zr (2), Hf (3), R = H; and M = Zr (4), Hf (5), R = Bu] were prepared and characterized by 77Se NMR spectroscopy and the crystal structures of 1–3 and 5 were determined by single-crystal X-ray diffraction. The crystal structure of 4 is known and the complex is iso- morphous with 5. 1–5 form mutually similar macrocyclic tetranuclear complexes in which the alternating Received 23rd January 2018, Fe(C5H4Se)2 and M(C5H4R)2 centers are linked by selenium bridges. The thermogravimetric analysis (TGA) Accepted 9th March 2018 of 1–3 under a helium atmosphere indicated that the complexes undergo a two-step decomposition DOI: 10.1039/c8dt00300a upon heating. The final products were identified using powder X-ray diffraction as FexMSe2, indicating Creative Commons Attribution-NonCommercial 3.0 Unported Licence. rsc.li/dalton their potential as single-source precursors for functional materials. Introduction been widely developed after the discovery that these materials may be used as precursors for semiconducting binary metal Transition metal dichalcogenides (TMDs) have been widely chalcogenides.13 Extension of this area to include various studied over the years due to their ability to intercalate species metallocene dichalcogenides has been possible because of the with apparent implications in catalytic and biological development of equipment for air-sensitive synthesis. -

WKS 4.1 - the Scientists (1 Page)
Pre-AP Chemistry Unit 4 HW Packet Name _____________________________ WKS 4.1 - The Scientists (1 page) The List of Scientists (Can be used more than once) Johann Dobereiner John Newlands Dimitri Mendeleev Henry Moseley Glenn Seaborg John Dalton J.J. Thomson Ernest Rutherford Neils Bohr Erwin Schrodinger Write the name of the scientist by his contribution….. 1. Who performed the Cathode Ray Tube Experiment? 2. Electrons were in energy levels. The further an electron was from the nucleus, the higher its energy. 3. Arranged the elements according to atomic number 4. Recognized the relationship between properties of “triads” of elements and their atomic mass. 5. Postulated the quantum (wave) mechanical model 6. The atom as mostly space and that all of the positive charge was located in a very small central nucleus 7. Synthesized transuranic elements 8. Published a version of the periodic table that was arranged according to atomic mass. wisely included blanks for unknown elements that I predicted would be discovered 9. Elements were arranged according to atomic mass that every 8th element seemed to have similar properties 10. “Billiard Ball” theory 11. Discovered that electrons were negative? 12. Who developed the first theory of the atom? 13. Who developed the planetary model? 14. Who performed the Gold Foil Experiment? 15. Who developed the Plum Pudding Model? 16. List the conclusions made from the Gold Foil Experiment. 90% of a worksheet must be completed to earn credit for that worksheet! Page 1 of 9 Pre-AP Chemistry Unit 4 HW Packet Name _____________________________ WKS 4.2 – Element Search (1 page) Use a highlighter or a pen to find the full names of the following elements. -

Lithium Alkaline Earth Tetrelides of the Type Li Aett (Ae = Ca, Ba, Tt = Si
Z. Naturforsch. 2017; 72(11)b: 847–853 Dominik Stoiber, Matej Bobnar, Peter Höhn and Rainer Niewa* Lithium alkaline earth tetrelides of the type Li2AeTt (Ae = Ca, Ba, Tt = Si, Ge, Sn, Pb): synthesis, crystal structures and physical properties https://doi.org/10.1515/znb-2017-0103 Heusler-type (MnCu2Al) structure with occupancy of octa- Received June 28, 2017; accepted July 20, 2017 hedral holes by Li and mixed Li/Mg occupancy in tetrahe- dral holes of the fcc packing of Tt. More recently, Li MgSi Abstract: Single crystals of the compounds Li AeTt 2 2 was reported [3, 4] with ordered lithium and magnesium (Ae = Ca, Ba, Tt = Si, Ge, Sn, Pb) were grown in reactive arrangements leading to lower crystallographic symmetry. lithium melts in sealed tantalum ampoules from an equi- One model comprises a 2 × 2 × 2 superstructure, the second molar ratio of the alkaline earth metal and the respective a more complicated order. The compounds Li CaSn and group 4 element. All compounds, with the exception of 2 Li CaPb also exhibit a Heusler-type crystal structure, but Li CaSn and Li CaPb, are isotypic and crystallize in an 2 2 2 in contrast to the Mg containing phases with an ordered orthorhombic unit cell (space group Pmmn, no. 59). The arrangement of fully occupied sites with Li in tetrahedral crystal structure can be characterized as superimposed and Ca in octahedral coordination by Tt, due to the larger corrugated networks of Li Tt connected by calcium or 2 difference in ionic radii compared to the magnesium com- barium atoms within the third dimension. -

WKS 4.1 - the Scientists (1 Page)
Pre-AP Chemistry Unit 4 Practice Packet Name _____________________________ WKS 4.1 - The Scientists (1 page) The List of Scientists (Can be used more than once!!!) Johann Dobereiner John Newlands Dimitri Mendeleev Henry Moseley Glenn Seaborg John Dalton J.J. Thomson Ernest Rutherford Neils Bohr Erwin Schrodinger Write the name of the scientist by his contribution….. 1. Who performed the Cathode Ray Tube Experiment? 2. Electrons were in energy levels. The further an electron was from the nucleus, the higher its energy. 3. Arranged the elements according to atomic number 4. Recognized the relationship between properties of “triads” of elements and their atomic mass. 5. Postulated the quantum (wave) mechanical model 6. The atom as mostly space and that all of the positive charge was located in a very small central nucleus 7. Synthesized transuranic elements 8. Published a version of the periodic table that was arranged according to atomic mass. wisely included blanks for unknown elements that I predicted would be discovered 9. Elements were arranged according to atomic mass that every 8th element seemed to have similar properties 10. “Billiard Ball” theory 11. Discovered that electrons were negative? 12. Who developed the first theory of the atom? 13. Who developed the planetary model? 14. Who performed the Gold Foil Experiment? 15. Who developed the Plum Pudding Model? 16. List the conclusions made from the Gold Foil Experiment. Page 1 of 9 Pre-AP Chemistry Unit 4 Practice Packet Name _____________________________ WKS 4.2 – Element Search (1 page) Use a highlighter or a pen to find the full names of the following elements. -

Macrocycles Containing 1, 1′-Ferrocenyldiselenolato Ligands
Dalton Transactions View Article Online PAPER View Journal | View Issue Macrocycles containing 1,1’-ferrocenyldiselenolato ligands Cite this: Dalton Trans., 2018, 47, 5415 on group 4 metallocenes† Clara Sanchez-Perez,a,b Caroline E. Knapp, b Minna M. Karjalainen,a Raija Oilunkaniemi,a Claire J. Carmalt *b and Risto S. Laitinen *a 5 5 t Macrocyclic [Fe(η -C5H4Se)2M(η -C5H4R)2]2 [M = Ti (1), Zr (2), Hf (3), R = H; and M = Zr (4), Hf (5), R = Bu] were prepared and characterized by 77Se NMR spectroscopy and the crystal structures of 1–3 and 5 were determined by single-crystal X-ray diffraction. The crystal structure of 4 is known and the complex is iso- morphous with 5. 1–5 form mutually similar macrocyclic tetranuclear complexes in which the alternating Received 23rd January 2018, Fe(C5H4Se)2 and M(C5H4R)2 centers are linked by selenium bridges. The thermogravimetric analysis (TGA) Accepted 9th March 2018 of 1–3 under a helium atmosphere indicated that the complexes undergo a two-step decomposition DOI: 10.1039/c8dt00300a upon heating. The final products were identified using powder X-ray diffraction as FexMSe2, indicating Creative Commons Attribution-NonCommercial 3.0 Unported Licence. rsc.li/dalton their potential as single-source precursors for functional materials. Introduction been widely developed after the discovery that these materials may be used as precursors for semiconducting binary metal Transition metal dichalcogenides (TMDs) have been widely chalcogenides.13 Extension of this area to include various studied over the years due to their ability to intercalate species metallocene dichalcogenides has been possible because of the with apparent implications in catalytic and biological development of equipment for air-sensitive synthesis. -

Studies of Fundamental Properties of Rutherfordium (Element 104) Using Organic Complexing Agents
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title Studies of Fundamental Properties of Rutherfordium (element 104) using Organic Complexing Agents Permalink https://escholarship.org/uc/item/8h092910 Author Czerwinski, K.R. Publication Date 1992-04-01 eScholarship.org Powered by the California Digital Library University of California LBL--32233 DE92 016925 Studies of Fundamental Properties of Rutherfordium a, (Element 104) Using Organic Complexing Agents Kenneth R, Czerwinski Ph.D. Thesis Department of Chemistr7 University of California and Nuclear Science Division Lawrence Berkeley Laboratory University of California _, Berkeley, CA 94720 Apill 1992 q "Fhi:¢work was supported by the Dirt..ctor,Office of Energy Research, Division of Nuclear Physics of the Office.of High Energy and Nuclear Physics oi the U,S. D.ep_tment of Energy under Contract No. I)E-AC()3-76SFO0098. t_;t'*_"iv_|':_ i i g,/.;i..,i_,'Liiv,ii c.._ "' '" .... a Studies of Fundamental Properties of Rutherfordium (Element 104) Using ,Organic Complexing Agents by Ken R. Czerwinski u, Abstract . Chemical properties of rutherfordium (Rf) have been investigated with the organic ligands triisooctylamine (TIOA), tributylphosphate (TBP), and thenoyltrifluoroacetone (TTA). The TIOA studies showed that Rf behaves differently than Th and Eu and most similarly to Zr, only Zr and Rf extract from 12 M HCI. This result is further evidence that Rf is a Group 4 element. Studies with TBP showed that Rf chemical behavior differed from the other Group 4 elements. The extractior_ by TBP at different chloride concentrations showed that Rf at times behaves more like Pu 4+ than Zr or Hf. -
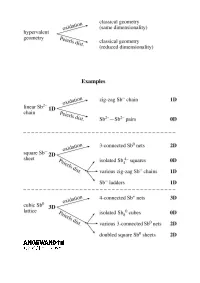
Extending the Zintl ± Klemm Concept to Nonclassical Electron-Rich Networks
REVIEWS Hypervalent Bonding in One, Two, and Three Dimensions: Extending the Zintl ± Klemm Concept to Nonclassical Electron-Rich Networks Garegin A. Papoian* and Roald Hoffmann* We construct a theory for electron-rich s-p mixing (or its absence) plays a structure types, seemingly unrelated to polyanionic networks in the interme- critical role in the propensity for pair- each other, may be understood using tallic compounds of heavy late main ing and, in general, in determining the the unifying concept of electron-rich group elements, building a bonding geometrical and electronic structure of multicenter bonding. Antimonides, framework that makes a connection these phases. Hypervalent bonding which are explored in great detail in to well-understood hypervalent bond- goes along with the relative absence the current work, conform particularly ing in small molecules such as XeF4, of significant s-p interaction; there is a well to the set of electron counting À XeF2 , and I3 . What we do is similar in continuum of such mixing, but also a rules for electron-rich nonclassical net- spirit to the analogy between the significant difference between the sec- works. Some deviation up and down Zintl ± Klemm treatment of classical ond-row and heavier elements. We from the ideal electron count is ex- polyanionic networks and the octet attribute the existence of undistorted hibited by known stannides and tellur- rule for molecules. We show that the metallic networks of the latter ele- ides. We can also make sense of the optimal electron count for a linear ments to diminished s-p mixing, which bonding in substantially more compli- chain of a heavy main group element in turn is due to the contraction of less- cated alloys, including La12Mn2Sb30 and is seven electrons per atom, six elec- screened s orbitals relative to p orbitals Tl4SnTe3 . -
The Electronic Structure of the Atoms of Five Elements Are Shown in the Figure Below. the Letters Are Not the Symbols of The
www.tutorzone.co.uk The electronic structure of the atoms of five elements are shown in the figure below. 1 The letters are not the symbols of the elements. Choose the element to answer the question. Each element can be used once, more than once or not at all. Use the periodic table to help you. (a) Which element is hydrogen? Tick one box. A B C D E (1) (b) Which element is a halogen? Tick one box. A B C D E (1) (c) Which element is a metal in the same group of the periodic table as element A? Tick one box. A B C D E (1) Page 1 of 99 www.tutorzone.co.uk (d) Which element exists as single atoms? Tick one box. A B C D E (1) (e) There are two isotopes of element A. Information about the two isotopes is shown in the table below. Mass number of the isotope 6 7 Percentage abundance 92.5 7.5 Use the information in the table above above to calculate the relative atomic mass of element A. Give your answer to 2 decimal places. ............................................................................................................................. ............................................................................................................................. ............................................................................................................................. ............................................................................................................................. ............................................................................................................................