The Raxml V8.2.X Manual
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 A) Login to the System B) Use the Appropriate Command to Determine Your Login Shell C) Use the /Etc/Passwd File to Verify the Result of Step B
CSE ([email protected] II-Sem) EXP-3 1 a) Login to the system b) Use the appropriate command to determine your login shell c) Use the /etc/passwd file to verify the result of step b. d) Use the ‘who’ command and redirect the result to a file called myfile1. Use the more command to see the contents of myfile1. e) Use the date and who commands in sequence (in one line) such that the output of date will display on the screen and the output of who will be redirected to a file called myfile2. Use the more command to check the contents of myfile2. 2 a) Write a “sed” command that deletes the first character in each line in a file. b) Write a “sed” command that deletes the character before the last character in each line in a file. c) Write a “sed” command that swaps the first and second words in each line in a file. a. Log into the system When we return on the system one screen will appear. In this we have to type 100.0.0.9 then we enter into editor. It asks our details such as Login : krishnasai password: Then we get log into the commands. bphanikrishna.wordpress.com FOSS-LAB Page 1 of 10 CSE ([email protected] II-Sem) EXP-3 b. use the appropriate command to determine your login shell Syntax: $ echo $SHELL Output: $ echo $SHELL /bin/bash Description:- What is "the shell"? Shell is a program that takes your commands from the keyboard and gives them to the operating system to perform. -
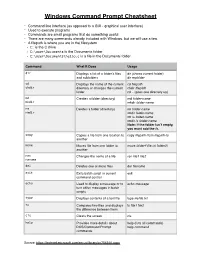
Windows Command Prompt Cheatsheet
Windows Command Prompt Cheatsheet - Command line interface (as opposed to a GUI - graphical user interface) - Used to execute programs - Commands are small programs that do something useful - There are many commands already included with Windows, but we will use a few. - A filepath is where you are in the filesystem • C: is the C drive • C:\user\Documents is the Documents folder • C:\user\Documents\hello.c is a file in the Documents folder Command What it Does Usage dir Displays a list of a folder’s files dir (shows current folder) and subfolders dir myfolder cd Displays the name of the current cd filepath chdir directory or changes the current chdir filepath folder. cd .. (goes one directory up) md Creates a folder (directory) md folder-name mkdir mkdir folder-name rm Deletes a folder (directory) rm folder-name rmdir rmdir folder-name rm /s folder-name rmdir /s folder-name Note: if the folder isn’t empty, you must add the /s. copy Copies a file from one location to copy filepath-from filepath-to another move Moves file from one folder to move folder1\file.txt folder2\ another ren Changes the name of a file ren file1 file2 rename del Deletes one or more files del filename exit Exits batch script or current exit command control echo Used to display a message or to echo message turn off/on messages in batch scripts type Displays contents of a text file type myfile.txt fc Compares two files and displays fc file1 file2 the difference between them cls Clears the screen cls help Provides more details about help (lists all commands) DOS/Command Prompt help command commands Source: https://technet.microsoft.com/en-us/library/cc754340.aspx. -

Don't Trust Traceroute (Completely)
Don’t Trust Traceroute (Completely) Pietro Marchetta, Valerio Persico, Ethan Katz-Bassett Antonio Pescapé University of Southern California, CA, USA University of Napoli Federico II, Italy [email protected] {pietro.marchetta,valerio.persico,pescape}@unina.it ABSTRACT In this work, we propose a methodology based on the alias resolu- tion process to demonstrate that the IP level view of the route pro- vided by traceroute may be a poor representation of the real router- level route followed by the traffic. More precisely, we show how the traceroute output can lead one to (i) inaccurately reconstruct the route by overestimating the load balancers along the paths toward the destination and (ii) erroneously infer routing changes. Categories and Subject Descriptors C.2.1 [Computer-communication networks]: Network Architec- ture and Design—Network topology (a) Traceroute reports two addresses at the 8-th hop. The common interpretation is that the 7-th hop is splitting the traffic along two Keywords different forwarding paths (case 1); another explanation is that the 8- th hop is an RFC compliant router using multiple interfaces to reply Internet topology; Traceroute; IP alias resolution; IP to Router to the source (case 2). mapping 1 1. INTRODUCTION 0.8 Operators and researchers rely on traceroute to measure routes and they assume that, if traceroute returns different IPs at a given 0.6 hop, it indicates different paths. However, this is not always the case. Although state-of-the-art implementations of traceroute al- 0.4 low to trace all the paths -

Forest Quickstart Guide for Linguists
Forest Quickstart Guide for Linguists Guido Vanden Wyngaerd [email protected] June 28, 2020 Contents 1 Introduction 1 2 Loading Forest 2 3 Basic Usage 2 4 Adjusting node spacing 4 5 Triangles 7 6 Unlabelled nodes 9 7 Horizontal alignment of terminals 10 8 Arrows 11 9 Highlighting 14 1 Introduction Forest is a package for drawing linguistic (and other) tree diagrams de- veloped by Sašo Živanović. This manual provides a quickstart guide for linguists with just the essential things that you need to get started. More 1 extensive documentation is available from the CTAN-archive. Forest is based on the TikZ package; more information about its commands, in par- ticular those controlling the appearance of the nodes, the arrows, and the highlighting can be found in the TikZ documentation. 2 Loading Forest In your preamble, put \usepackage[linguistics]{forest} The linguistics option makes for nice trees, in which the branches meet above the two nodes that they join; it will also align the example number (provided by linguex) with the top of the tree: (1) CP C IP I VP V NP 3 Basic Usage Forest uses a familiar labelled brackets syntax. The code below will out- put the tree in (1) above (\ex. requires the linguex package and provides the example number): \ex. \begin{forest} [CP[C][IP[I][VP[V][NP]]]] \end{forest} Forest will parse the above code without problem, but you are likely to soon get lost in your labelled brackets with more complicated trees if you write the code this way. The better alternative is to arrange the nodes over multiple lines: 2 \ex. -

What Is UNIX? the Directory Structure Basic Commands Find
What is UNIX? UNIX is an operating system like Windows on our computers. By operating system, we mean the suite of programs which make the computer work. It is a stable, multi-user, multi-tasking system for servers, desktops and laptops. The Directory Structure All the files are grouped together in the directory structure. The file-system is arranged in a hierarchical structure, like an inverted tree. The top of the hierarchy is traditionally called root (written as a slash / ) Basic commands When you first login, your current working directory is your home directory. In UNIX (.) means the current directory and (..) means the parent of the current directory. find command The find command is used to locate files on a Unix or Linux system. find will search any set of directories you specify for files that match the supplied search criteria. The syntax looks like this: find where-to-look criteria what-to-do All arguments to find are optional, and there are defaults for all parts. where-to-look defaults to . (that is, the current working directory), criteria defaults to none (that is, select all files), and what-to-do (known as the find action) defaults to ‑print (that is, display the names of found files to standard output). Examples: find . –name *.txt (finds all the files ending with txt in current directory and subdirectories) find . -mtime 1 (find all the files modified exact 1 day) find . -mtime -1 (find all the files modified less than 1 day) find . -mtime +1 (find all the files modified more than 1 day) find . -

NETSTAT Command
NETSTAT Command | NETSTAT Command | Use the NETSTAT command to display network status of the local host. | | ┌┐────────────── | 55──NETSTAT─────6─┤ Option ├─┴──┬────────────────────────────────── ┬ ─ ─ ─ ────────────────────────────────────────5% | │┌┐───────────────────── │ | └─(──SELect───6─┤ Select_String ├─┴ ─ ┘ | Option: | ┌┐─COnn────── (1, 2) ──────────────── | ├──┼─────────────────────────── ┼ ─ ──────────────────────────────────────────────────────────────────────────────┤ | ├─ALL───(2)──────────────────── ┤ | ├─ALLConn─────(1, 2) ────────────── ┤ | ├─ARp ipaddress───────────── ┤ | ├─CLients─────────────────── ┤ | ├─DEvlinks────────────────── ┤ | ├─Gate───(3)─────────────────── ┤ | ├─┬─Help─ ┬─ ───────────────── ┤ | │└┘─?──── │ | ├─HOme────────────────────── ┤ | │┌┐─2ð────── │ | ├─Interval─────(1, 2) ─┼───────── ┼─ ┤ | │└┘─seconds─ │ | ├─LEVel───────────────────── ┤ | ├─POOLsize────────────────── ┤ | ├─SOCKets─────────────────── ┤ | ├─TCp serverid───(1) ─────────── ┤ | ├─TELnet───(4)───────────────── ┤ | ├─Up──────────────────────── ┤ | └┘─┤ Command ├───(5)──────────── | Command: | ├──┬─CP cp_command───(6) ─ ┬ ────────────────────────────────────────────────────────────────────────────────────────┤ | ├─DELarp ipaddress─ ┤ | ├─DRop conn_num──── ┤ | └─RESETPool──────── ┘ | Select_String: | ├─ ─┬─ipaddress────(3) ┬ ─ ───────────────────────────────────────────────────────────────────────────────────────────┤ | ├─ldev_num─────(4) ┤ | └─userid────(2) ─── ┘ | Notes: | 1 Only ALLCON, CONN and TCP are valid with INTERVAL. | 2 The userid -

Introduction to Unix Shell
Introduction to Unix Shell François Serra, David Castillo, Marc A. Marti- Renom Genome Biology Group (CNAG) Structural Genomics Group (CRG) Run Store Programs Data Communicate Interact with each other with us The Unix Shell Introduction Interact with us Rewiring Telepathy Typewriter Speech WIMP The Unix Shell Introduction user logs in The Unix Shell Introduction user logs in user types command The Unix Shell Introduction user logs in user types command computer executes command and prints output The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off shell The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output -

CS101 Lecture 8
What is a program? What is a “Window Manager” ? What is a “GUI” ? How do you navigate the Unix directory tree? What is a wildcard? Readings: See CCSO’s Unix pages and 8-2 Applications Unix Engineering Workstations UNIX- operating system / C- programming language / Matlab Facilitate machine independent program development Computer program(software): a sequence of instructions that tells the computer what to do. A program takes raw data as input and produces information as output. System Software: • Operating Systems Unix,Windows,MacOS,VM/CMS,... • Editor Programs gedit, pico, vi Applications Software: • Translators and Interpreters gcc--gnu c compiler matlab – interpreter • User created Programs!!! 8-4 X-windows-a Window Manager and GUI(Graphical User Interface) Click Applications and follow the menus or click on an icon to run a program. Click Terminal to produce a command line interface. When the following slides refer to Unix commands it is assumed that these are entered on the command line that begins with the symbol “>” (prompt symbol). Data, information, computer instructions, etc. are saved in secondary storage (auxiliary storage) in files. Files are collected or organized in directories. Each user on a multi-user system has his/her own home directory. In Unix users and system administrators organize files and directories in a hierarchical tree. secondary storage Home Directory When a user first logs in on a computer, the user will be “in” the users home directory. To find the name of the directory type > pwd (this is an acronym for print working directory) So the user is in the directory “gambill” The string “/home/gambill” is called a path. -

Trees, Binary Search Trees, Heaps & Applications Dr. Chris Bourke
Trees Trees, Binary Search Trees, Heaps & Applications Dr. Chris Bourke Department of Computer Science & Engineering University of Nebraska|Lincoln Lincoln, NE 68588, USA [email protected] http://cse.unl.edu/~cbourke 2015/01/31 21:05:31 Abstract These are lecture notes used in CSCE 156 (Computer Science II), CSCE 235 (Dis- crete Structures) and CSCE 310 (Data Structures & Algorithms) at the University of Nebraska|Lincoln. This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License 1 Contents I Trees4 1 Introduction4 2 Definitions & Terminology5 3 Tree Traversal7 3.1 Preorder Traversal................................7 3.2 Inorder Traversal.................................7 3.3 Postorder Traversal................................7 3.4 Breadth-First Search Traversal..........................8 3.5 Implementations & Data Structures.......................8 3.5.1 Preorder Implementations........................8 3.5.2 Inorder Implementation.........................9 3.5.3 Postorder Implementation........................ 10 3.5.4 BFS Implementation........................... 12 3.5.5 Tree Walk Implementations....................... 12 3.6 Operations..................................... 12 4 Binary Search Trees 14 4.1 Basic Operations................................. 15 5 Balanced Binary Search Trees 17 5.1 2-3 Trees...................................... 17 5.2 AVL Trees..................................... 17 5.3 Red-Black Trees.................................. 19 6 Optimal Binary Search Trees 19 7 Heaps 19 -

Mkdir / MD Command :- This Command Is Used to Create the Directory in the Dos
Department Of Computer Application (BBA) Dr. Rakesh Ranjan BCA Sem - 2 Operating system Mkdir / MD Command :- this command is used to create the directory in the dos . we can also create subdirectory within the directory by this command . we have to give path of the proposed directory to create. Syntax : c:\> mkdir abc This command create the directory abc on the c: drive C:\> md abc\cde It create subdirectory cde under abc directory C:\> md abc\pqr C:\md abc\cde\a C:\ md abc\cde\b C:\md abc\pqr\a C:\ md abc\pqr\b C:\md abc\pqr\a\c C:\ md abc\pqr\a\d The structure of the above created directory may be shown as ABC CDE PQR A B A B C D Tree command :- this command in dos is used to display the structure of the directory and subdirectory in DOS. C:\> tree This command display the structure of c: Drive C:\> tree abc It display the structure of abc directory on the c: drive Means that tree command display the directory structure of the given directory of given path. RD Command :- RD stands for Remove directory command . this command is used to remove the directory from the disk . it should be noted that directory which to be removed must be empty otherwise , can not be removed . Syntax :- c:\ rd abc It display error because , abc is not empty C:\ rd abc\cde It also display error because cde is not empty C:\ rd abc\cde\a It works and a directory will remove from the disk C:\ rd abc\cde\b It will remove the b directory C:\rd abc\cde It will remove the cde directory because cde is now empty. -

UNIX (Solaris/Linux) Quick Reference Card Logging in Directory Commands at the Login: Prompt, Enter Your Username
UNIX (Solaris/Linux) QUICK REFERENCE CARD Logging In Directory Commands At the Login: prompt, enter your username. At the Password: prompt, enter ls Lists files in current directory your system password. Linux is case-sensitive, so enter upper and lower case ls -l Long listing of files letters as required for your username, password and commands. ls -a List all files, including hidden files ls -lat Long listing of all files sorted by last Exiting or Logging Out modification time. ls wcp List all files matching the wildcard Enter logout and press <Enter> or type <Ctrl>-D. pattern Changing your Password ls dn List files in the directory dn tree List files in tree format Type passwd at the command prompt. Type in your old password, then your new cd dn Change current directory to dn password, then re-enter your new password for verification. If the new password cd pub Changes to subdirectory “pub” is verified, your password will be changed. Many systems age passwords; this cd .. Changes to next higher level directory forces users to change their passwords at predetermined intervals. (previous directory) cd / Changes to the root directory Changing your MS Network Password cd Changes to the users home directory cd /usr/xx Changes to the subdirectory “xx” in the Some servers maintain a second password exclusively for use with Microsoft windows directory “usr” networking, allowing you to mount your home directory as a Network Drive. mkdir dn Makes a new directory named dn Type smbpasswd at the command prompt. Type in your old SMB passwword, rmdir dn Removes the directory dn (the then your new password, then re-enter your new password for verification. -

System Analysis and Tuning Guide System Analysis and Tuning Guide SUSE Linux Enterprise Server 15 SP1
SUSE Linux Enterprise Server 15 SP1 System Analysis and Tuning Guide System Analysis and Tuning Guide SUSE Linux Enterprise Server 15 SP1 An administrator's guide for problem detection, resolution and optimization. Find how to inspect and optimize your system by means of monitoring tools and how to eciently manage resources. Also contains an overview of common problems and solutions and of additional help and documentation resources. Publication Date: September 24, 2021 SUSE LLC 1800 South Novell Place Provo, UT 84606 USA https://documentation.suse.com Copyright © 2006– 2021 SUSE LLC and contributors. All rights reserved. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.2 or (at your option) version 1.3; with the Invariant Section being this copyright notice and license. A copy of the license version 1.2 is included in the section entitled “GNU Free Documentation License”. For SUSE trademarks, see https://www.suse.com/company/legal/ . All other third-party trademarks are the property of their respective owners. Trademark symbols (®, ™ etc.) denote trademarks of SUSE and its aliates. Asterisks (*) denote third-party trademarks. All information found in this book has been compiled with utmost attention to detail. However, this does not guarantee complete accuracy. Neither SUSE LLC, its aliates, the authors nor the translators shall be held liable for possible errors or the consequences thereof. Contents About This Guide xii 1 Available Documentation xiii