A Novel Prognostic DNA Methylation Panel for Colorectal Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

OSAC Encourages Travelers to Use This Report to Gain Baseline Knowledge of Security Conditions in Taiwan
Taiwan 2020 Crime & Safety Report This is an annual report produced in conjunction with the Regional Security Office at the American Institute in Taiwan (AIT) in Taipei. The U.S. maintains unofficial relations with the people of Taiwan through AIT, a private nonprofit corporation, which performs citizen and consular services similar to those at diplomatic posts. OSAC encourages travelers to use this report to gain baseline knowledge of security conditions in Taiwan. For more in-depth information, review OSAC’s Taiwan page for original OSAC reporting, consular messages, and contact information, some of which may be available only to private- sector representatives with an OSAC password. Travel Advisory The current U.S. Department of State Travel Advisory at the date of this report’s publication assesses Taiwan at Level 1, indicating travelers should exercise normal precautions. Review OSAC’s report, Understanding the Consular Travel Advisory System. Overall Crime and Safety Situation Crime Threats The U.S. Department of State has assessed Taipei as being a LOW-threat location for crime. There is extensive CCTV coverage throughout Taiwan (30,000 cameras in Taipei, 25,000 in Kaohsiung, and 35,000 in New Taipei City). The network of security cameras at traffic intersections, commercial establishments, and public areas of hotels and residential buildings means that authorities can monitor and/or record most activity occurring outside of the home. The extensive coverage plays a significant role in deterring the majority of criminal activity, and most streets in Taiwan are generally safe. Pickpockets can be a problem in crowded areas, especially at various night markets. -

*THJ0423024* Page 1 of 7 Intertek Testing Services Taiwan Ltd
Test Report Number : TWNC00423024 Applicant: Formosa Plastics Corporation Date : Mar 19, 2015 Technical department plastics divison 100,Suei-Guan Road,Jen-Wu Hsiang, Kaohsiung County Sample Description: One (1) group of submitted samples said to be : Sample Description : M-81 Date Sample Received : Feb 05, 2015 Date Test Started : Feb 09, 2015 Test Conducted : As requested by the applicant, for details please refer to attached pages. Conclusion: Tested Sample Standard Result Submitted Samples Washington Chemicals of High Concern to Children (CHCC) See Test Content (as Contaminant) Conducted - As per Reporting Rule (Chapter 173-334 WAC) under Children's Safe Products Act (CSPA) - Chapter 70.240 (RCW) Remark: As per clause (1)(b) of Washington Adminstration Code (WAC) 173-334-080 of chapter 173-334 children's safe products -reporting rule, each chemical on the CHCC list that is a contaminant present in a product component must be reported at concentration above 100 ppm. The manufacturer must include the product component in the notice to the Washington state department of Ecology unless the manufacturer had in place a manufacturing control program and exercised due diligence to minimize the presence of the contaminant in the component. Authorized by: On Behalf of Intertek Testing Services Taiwan Limited K. Y. Liang Director *THJ0423024* Page 1 of 7 Intertek Testing Services Taiwan Ltd. 8F., No. 423, Ruiguang Rd., Neihu District, Taipei 11492, Taiwan, R.O.C. 全國公證檢驗股份有限公司 11492 台北市內湖區瑞光路 423 號 8 樓 Tel: (+886-2) 6602-2888.2797-8885 Fax: (+886-2) 6602-2410 Test Report Number : TWNC00423024 Test Conducted Washington Chemicals of High Concern to Children (CHCC) Content (as Contaminant) By Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES), Gas Chromatography-Mass Spectrometer linked with Headspace (GC-MS linked with Headspace), Gas Chromatography-Mass Spectrometer (GC-MS), High Performance Liquid Chromatography-Photodiode Array Detector (HPLC-DAD), and Liquid Chromatography /Tandem Mass Spectrometer (LC-MS-MS) analysis. -

Analysis of Residential and Auto Break-In Records in Taipei City
Portland State University PDXScholar Engineering and Technology Management Student Projects Engineering and Technology Management Winter 2018 Analysis of Residential and Auto Break-in Records in Taipei City Afnan Althoupety Portland State University Aishwarya Joy Portland State University Juchun Cheng Portland State University Priyanka Patil Portland State University Tejas Deshpande Portland State University Follow this and additional works at: https://pdxscholar.library.pdx.edu/etm_studentprojects Part of the Applied Statistics Commons, and the Social Control, Law, Crime, and Deviance Commons Let us know how access to this document benefits ou.y Citation Details Althoupety, Afnan; Joy, Aishwarya; Cheng, Juchun; Patil, Priyanka; and Deshpande, Tejas, "Analysis of Residential and Auto Break-in Records in Taipei City" (2018). Engineering and Technology Management Student Projects. 1943. https://pdxscholar.library.pdx.edu/etm_studentprojects/1943 This Project is brought to you for free and open access. It has been accepted for inclusion in Engineering and Technology Management Student Projects by an authorized administrator of PDXScholar. Please contact us if we can make this document more accessible: [email protected]. Analysis of Residential and Auto Break-in Records in Taipei City Course: ETM 538 Term: Winter 2018 Instructors: Daniel Sagalowicz, Mike Freiling Date: 03/09/2018 Authors:Afnan Althoupety , Aishwarya Joy , Juchun Cheng , Priyanka Patil , Tejas Deshpande 1 1. Introduction Taipei City is the capital of Taiwan. It has population of 2.7 million living in the city area of 271 km2 (104 mi2). There are totally 12 administrative districts in this city. To maintain the safety of the city, Taipei City Police Bureau has arranged regular patrol routes with focus on the high-risk area where residential and auto break-in occurs. -

Detecting the Issues of Population Aging by Using Arima Model
ICIC Express Letters Part B: Applications ICIC International ⃝c 2019 ISSN 2185-2766 Volume 10, Number 1, January 2019 pp. 39{45 DETECTING THE ISSUES OF POPULATION AGING BY USING ARIMA MODEL Dian-Fu Chang1;∗, Yi Hsing Lin2 and Fong-Yee Nyeu3 1Graduate Institute of Educational Policy and Leadership 2Doctoral Program of Educational Leadership and Technology Management Tamkang University No. 151, Yingzhuan Road, Tamsui District, New Taipei City 25137, Taiwan ∗Corresponding author: [email protected]; [email protected] 3Office of International and Cross-Strait Affair University of Kang Ning No. 137, Kang Ning Road, Neihu District, Taipei City 11485, Taiwan [email protected] Received June 2018; accepted September 2018 Abstract. Population aging is poised to become one of the most significant social trans- formations of the twenty-first century. Perceiving the rapidly increasing population aging, this study selected Taiwan as a target to tackle the issue. Previous studies have warned the drawbacks of composite indicators dealing with aging issues; therefore, this study tries to find an alternative way to fit series data in an aging society. To achieve the specific purposes, the study aims to create appropriate indices to further interpret the issues of population aging. The time series analysis was carried out in this study by using the Minitab⃝R statistical package. The result of the ARIMA model reveals the increasing of over 65 year-old adults in Taiwan will reach up to a new high in next decade. The po- tential support ratio will decline speedily. Realizing the gap, the suggestions are provided for the policy makers to deal with the issues more effectively. -

Brief Introduction of Employment Service Resources
MRT map for each Employment Service Station Taipei City Employment Services Oce 8th Floor, No.101, Bangka Boulevard, Beitou Employment Service Station Wanhua District, Taipei City Beitou 5th Floor, No. 30, Xinshih St. Beitou District, MRT "Longshan Temple Station" , Exit 2 Taipei City / MRT "Beitou Station"/ Tel: 02-2308-5230 or dial 1999 Nankang Software Park's Information Desk Brief Introduction of Beitou District Administration Building (within Taipei City) ext. 58999 Tel: 02-2898-1819 【Provides recruiting services only. Do not provide Employment Service Bangka Employment Service Station (re)-identication of employment and unemployment Neihu Employment Services Station services】 3rd Floor, No.101, Bangka Boulevard, 1st Floor, No. 3, Nankang Software Park Street, Resources Wanhua District, Taipei City 7th Floor, No. 99, Section 6, Minquan East Road, Nankang District, Taipei City / Joint Service MRT "Longshan Temple Station" , Exit 2 Neihu District, Taipei City/ Center, Area F, NankangSoftware Park / Tel: 02-2308-5231 Neihu District Administration Building MRT "Nangang Software Park Station" MRT "Wender Station" Tel: 02-2652-1322 OKWORK Tel: 02-2790-0399 8th Floor, No.101, Bangka Boulevard, Wanhua District, Taipei City Nankang MRT "Longshan Temple Station", Exit 2 Wende Software Tel: 02-2338-0277or dial 1999 Park Taipei Main Station Employment ( within Taipei City) ext. 58988 Service Desk Dinghao Employment And Senior Wanhua Employment Service Desk 【for inquiries only】 Services Station 【 1st Floor, No. 3, Beiping West Road, Taipei Only provides job searching and No. 77, Daan Road, Section 1, Daan District, recruiting services. Do not unemployment City / South West Area Employment Services Desk Taipei City, No. 1, East Metro Mall (re)-identication or vocational training MRT "Zhongxiao Dunhua Station" 】 MRT "Taipei Main Station" /Taipei Station consultation and recommendations Tel: 02-2740-0922 3rd Floor, No. -
![[カテゴリー]Location Type [スポット名]English Location Name [住所](https://docslib.b-cdn.net/cover/8080/location-type-english-location-name-1138080.webp)
[カテゴリー]Location Type [スポット名]English Location Name [住所
※IS12TではSSID"ilove4G"はご利用いただけません [カテゴリー]Location_Type [スポット名]English_Location_Name [住所]Location_Address1 [市区町村]English_Location_City [州/省/県名]Location_State_Province_Name [SSID]SSID_Open_Auth Misc Hi-Life-Jingrong Kaohsiung Store No.107 Zhenxing Rd. Qianzhen Dist. Kaohsiung City 806 Taiwan (R.O.C.) Kaohsiung CHT Wi-Fi(HiNet) Misc Family Mart-Yongle Ligang Store No.4 & No.6 Yongle Rd. Ligang Township Pingtung County 905 Taiwan (R.O.C.) Pingtung CHT Wi-Fi(HiNet) Misc CHT Fonglin Service Center No.62 Sec. 2 Zhongzheng Rd. Fenglin Township Hualien County Hualien CHT Wi-Fi(HiNet) Misc FamilyMart -Haishan Tucheng Store No. 294 Sec. 1 Xuefu Rd. Tucheng City Taipei County 236 Taiwan (R.O.C.) Taipei CHT Wi-Fi(HiNet) Misc 7-Eleven No.204 Sec. 2 Zhongshan Rd. Jiaoxi Township Yilan County 262 Taiwan (R.O.C.) Yilan CHT Wi-Fi(HiNet) Misc 7-Eleven No.231 Changle Rd. Luzhou Dist. New Taipei City 247 Taiwan (R.O.C.) Taipei CHT Wi-Fi(HiNet) Restaurant McDonald's 1F. No.68 Mincyuan W. Rd. Jhongshan District Taipei CHT Wi-Fi(HiNet) Restaurant Cobe coffee & beauty 1FNo.68 Sec. 1 Sanmin Rd.Banqiao City Taipei County Taipei CHT Wi-Fi(HiNet) Misc Hi-Life - Taoliang store 1F. No.649 Jhongsing Rd. Longtan Township Taoyuan County Taoyuan CHT Wi-Fi(HiNet) Misc CHT Public Phone Booth (Intersection of Sinyi R. and Hsinsheng South R.) No.173 Sec. 1 Xinsheng N. Rd. Dajan Dist. Taipei CHT Wi-Fi(HiNet) Misc Hi-Life-Chenhe New Taipei Store 1F. No.64 Yanhe Rd. Anhe Vil. Tucheng Dist. New Taipei City 236 Taiwan (R.O.C.) Taipei CHT Wi-Fi(HiNet) Misc 7-Eleven No.7 Datong Rd. -

Chapter 7 Special Notes Affiliates 1
Chapter 7 Special Notes Affiliates 1. Investment holding structure As of December 31, 2015 Taiwan Mobile 5.86% Co., Ltd. 2.56% 12% 100% 49.9% 100% Wealth Media Taipei New Taiwan Cellular Technology Co., Ltd. Horizon Co., Ltd. Co., Ltd. 44.38% 100% 100% 100% 100% 100% 100% 100% 100% 100% 100% Taiwan Win TV Global Wealth Global Forest Media Media TFN Media Taiwan Digital Taiwan Digital TCC Investment TWM Holding Teleservices & Taiwan Fixed momo.com Inc. Broadcasting Communications Technology Technology Co., Ltd. Service Co., Ltd. Co., Ltd. Co., Ltd. Technologies Co., Network Co., Ltd. Co., Ltd. Co., Ltd. Co., Ltd. Co., Ltd. Ltd. 6.83% 0.76% 100% 100% 100% 100% 76.26% 100% 100% 100% 92.38% 99.22% 100% 29.53%(Note) 100% 100% 100% 100% TCCI Fu Sheng Fuli Property Fuli Life Honest Asian Crown Globalview Phoenix Mangrove Yeong Jia Taiwan Kuro TWM TFN Union Travel Service Insurance Insurance Union Cable Investment and TT&T Holdings Agent Co., Development International Cable TV Co., Cable TV Co., Cable TV Co., Leh Cable TV Times Co., Communications TFN HK Ltd. Investment Co., Ltd. Agent Co., TV Co., Ltd. Development Co., Ltd. Ltd. Ltd. Co., Ltd. Co., Ltd. Ltd. Ltd. Ltd. Co., Ltd. Ltd. Co., Ltd. (Beijing) Ltd. Co., Ltd. 100% 100% Hongkong Yue Numerous Fortune Investment Kingdom Corp. Co., Ltd. 100% 100% Haobo Hong Kong Information Fubon Consulting Multimedia (Shenzhen) Technology Co., Co., Ltd. Ltd. 91.30% Fubon Gehua (Beijing) Enterprise Ltd. Note: 70.47% of shares are held under trustee accounts. 115 2. Affiliates’ profile December 31, 2015; Unit: NT$ (unless otherwise stated) Date of Name Address Paid-in capital Main business incorporation Taiwan Cellular Co., 7F, No. -

Sponsored by Ministry of Labor 臺北市就業服務處 Brief Introduction of Employment Service Resources Contact for Other
8th Floor, No.101, Bangka Boulevard, Wanhua District, Taipei City ● 02-2308-5230 or dial 1999( within Taipei City) ext. 58999 Danshui Xinyi Line MRT「Longshan Temple Station」, Exit 2 ● Bangnan Line ● Taipei City Employment Services Office Xindian Line ● 3rd Floor, No.101, Bangka Boulevard, Wenhu Line Wanhua District, Taipei City 02-2308-5231 MRT「Longshan Temple Station」, Exit 2 Bangka Employment Service Station Nangang No. 77, Daan Road, Section 1, Daan Software District, Taipei City, No. 30, East Provides recruiting services only. Shandao Temple /National Metro Mall Taiwan University Hospital 7th Floor, No. 99, Section 6, Minquan East Park Road, Neihu District, Taipei City 02-2740-0922 8th Floor, No.101, Bangka Boulevard, MRT「Zhongxiao Dunhua Station」 Beitou 02-2790-0399 Wanhua District, Taipei City Neihu District Administration Building 02-2338-0277 or dial 1999( within Taipei City) ext. 58988 MRT「Longshan Temple Station」, Exit 2 Taipei OKWORK Neihu Employment Services Station Taipei OKWORK counseling statio Only providesjob searching and recruiting services. Do not provide unemployment 3rd Floor, No. 251, Heping West Road, Sec. 3, For inquiries only No. 77, Daan Road, Section 1, Daan Wanhua District, Taipei City 1st Floor, No. 3, Beiping West Road, Taipei District, Taipei City, No. 30 & 33, East 02-2308-0236 City South WestArea Employment Services Wende Metro Mall MRT「Longshan Temple Station」, Exit 1 Desk 02-2740-0922 MRT「Zhongxiao Dunhua Station」 02-2381-1278 Wanhua Employment Service Desk MRT「Taipei Main Station」/ Taipei Station Services Station Taipei Main Station Employment Service Desk Stasiun Longshan Ximen Shandao Zhongxiao Nangang Temple Taipei Temple Fuxing Android iOS LINE 主視覺設計說明: National No. -
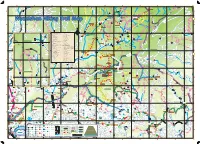
Wuzeishan Hiking Trail Map Lane 95 Bridge Connecting Point to the Small Picture on the Left
(Lane 43, Pingjing St, Industry Rd) Pinding Ancient Canal Ancient Pinding L a n S27 e Mt. Xinzhentou Dingshan 9 3 , (Xiajingque Industry Rd) Mt. Xingdui P i n g j i n Jingque Stream Jingque g Taozijiao Bridge S t ( N e Fengguizui Trail i l Mt. Neishuanxi i a Ciyun o nt C e I a (Xiaguoxi Industry Rd) Industry (Xiaguoxi Temple n ci nal d u s Stamp Pavilion An Wuzhishan Industry Rd t r Hi y g Qingfeng Pavilion n Tianweizi Bridge Wanxi Industry Rd ) d R i k d i g ng n Xiwan Rd Pi T r Pingjing St ail (Chencuo Industry Rd) S19 Neicuo Neicuo Bridge Shilin District, Taipei Sec 3, Zhishan Rd Xizhi District, New Taipei Daqitou Trail Chedengjiao Wuzeishan Hiking Trail Map Lane 95 Bridge Connecting point to the small picture on the left. Connecting point to the small picture ry Rd) Mt. Ewei ndust S I h u a St, n g g x i n g o ji Mt. Shuanxi u Mt. Wuzhishan g H i s in t P o r , i c Pingjing St (Pingxi Industry Rd) Industry (Pingxi St Pingjing Pingdeng S18 T 106 Shengren r (Lane 42, Pingjing St, Industry Rd) a Zhulin Trail Pingdingguchaun i Wanxi Industry Rd l (Lane Hiking Trail Entrance Waterfall N e Mt. Suiwei i Lane 371 s h (Lane 105, Pingjing St, Industry Rd) Industry St, Pingjing 105, (Lane u a n S Pingxi Trail t r e Mt. Meihua a Zhengtian Gongping Bridge m Temple Xiwan 3rd Bridge D6 Nature Trail Index of the Trails in Taipei S18 D7 The North Trail Shengren E5 Daluntoushan and Dalunweishan Shilin District, Taipei Xiwan Rd Yongzhen Trail Bridge Shengren Shanxi Touristic TaiPower Electricity Hiking Trail E6 Yuanming Temple Trail Bridge Farm Relay Station E5 Zhongshe Rd Branch Guandi Lutian Xinyi Rd B7 Daqitou Trail Temple Huaxin 2nd Rd E6 Bishan Rd Branch Mt. -

PAYS SECTION Taiwan Produits De La Pêche MISE EN GARDE Please
En vigueur depuis le: PAYS Taiwan 20/08/2021 00419 SECTION Produits de la pêche Date de publication 28/07/2007 Liste en vigueur Numéro Nom Ville Régions Activités Remarque Date de la demande d'agrément 2F30017 I-Mei Frozen Foods Company Limited Su-Ao Township Yilan County PP 2F30036 Hsien-Pin Frozen Foods Company Limited Su-Ao Township Yilan County PP Aq 2F30039 Hochico Marine Processing Corporation Su-Ao Township Yilan County PP 2FH0002 Land Young Foods Company Limited Wujie Township Yilan County PP 2FH0005 Jin Tzer Marine Products Company Limited Wujie Township Yilan County PP 2FH0009 JYY Fisheries Corp. Su'ao Yilan County PP 07/01/2011 2FH0013 Tong Ho Foods Industrial Company Limited Wu-Chei Hsiang Ilan Hsien PP 16/05/2011 6F3F001 Peak One International Corporation, Tainan Plant Guanmiau District Tainan City PP 21/01/2011 6F3F003 Shin Rong Business Co., Ltd. Chia-I-Hsien T'ai-wan PP Aq 13/08/2021 6FH0002 Tosei Sea Foods Company Limited Yi-Chu Hsiang, Chia Yi Hsien PP Aq 11/09/2013 6FH0006 Guarantee Liability Yun-Lin County Ko-Fwu Fishes Cooperation Kouhu Township Yunlin County PP Aq 6FH0008 Haw-Di-I Foods Company Limited Sinshih District Tainan City PP 6FH0012 Lian Ruey Enterprise Corporation Limited Annan District Tainan City PP 27/04/2010 7F30035 Tong Pao Frozen Food Company Limited Qiaotou District Kaohsiung City PP 7F30062 Shin Ho Sing Ocean Enterprise Company Limited Qianzhen District Kaohsiung City PP 25/01/2013 1 / 10 Liste en vigueur Numéro Nom Ville Régions Activités Remarque Date de la demande d'agrément 7F30075 Just Champion -

Directory of Head Office and Branches
Directory of Head Office and Branches 106 I. Domestic Business Units 120 Sec 1, Chongcing South Road, Jhongjheng District, Taipei City 10007, Taiwan (R.O.C.) P.O. Box 5 or 305 SWIFT: BKTWTWTP http://www.bot.com.tw TELEX 11201 TAIWANBK CODE OFFICE ADDRESS TELEPHONE FAX 0037 Department of 120 Sec 1, Chongcing South Road, Jhongjheng District, 02-23493399 02-23759708 Business ( I ) Taipei City 0059 Department of 120 Sec 1, Gueiyang Street, Jhongjheng District, 02-23615421 02-23751125 Public Treasury Taipei City 0071 Department of 49 Guancian Road, Jhongjheng District, Taipei City 02-23812949 02-23753800 Business ( II ) 0082 Department of 58 Sec 1, Chongcing South Road, Jhongjheng District, 02-23618030 02-23821846 Trusts Taipei City 0691 Offshore Banking 1F, 3 Baocing Road, Jhongjheng District, Taipei City 02-23493456 02-23894500 Branch 1850 Department of 4F, 120 Sec 1, Gueiyang Street, Jhongjheng District, 02-23494567 02-23893999 Electronic Banking Taipei City 1698 Department of 2F, 58 Sec 1, Chongcing South Road, Jhongjheng 02-23882188 02-23716159 Securities District, Taipei City 0093 Tainan Branch 155 Sec 1, Fucian Road, Central District, Tainan City 06-2160168 06-2160188 0107 Taichung Branch 140 Sec 1, Zihyou Road, West District, Taichung City 04-22224001 04-22224274 0118 Kaohsiung Branch 264 Jhongjheng 4th Road, Cianjin District, 07-2515131 07-2211257 Kaohsiung City 0129 Keelung Branch 16, YiYi Road, Jhongjheng District, Keelung City 02-24247113 02-24220436 0130 Chunghsin New 11 Guanghua Road, Jhongsing Village, Nantou City, 049-2332101 -

/ ID Last Update of Data Factory Production Process Supplier Name
Please use our contact form if you would like to have the list in another formate. VAUDE Supplier List 2019 1 of 6 pages Factory Production # / ID last update of data Supplier Name Process Country City Address #1 2017 Knitting, Dyeing AB Utenos Trikotazas Lithuania Utena J. Basanaviciaus str.122 #2 2017 Knitting A. Sampaio Portugal San Tirso Rua S. bento da Batalha, n° 76 Taipei World Trade Center Exhibition Hall, Room 2B35, No. 5 Hsin-Yi #3 2017 Zipperpuller Aicare Taiwan Taipei Road, Sec. 5 Cirpici, Veliefendi Yolu No: 49, #4 2018 Weaving, Dyeing Akin Türkei Istanbul Bakirkoy, Istanbul No.175,Lin Hao Road,HangZhou APEX (ZHE JIANG) Economic & Technological #5 2018 Dyeing TEXTILE CO., LTD China Hangzhou Development Area, 1.SEIREN CO.LTD Nitta 1st Plant 60-1, Nittazuka, 1 Chome, Fukui 910- 0067, Japan 2018 Converter ASF Japan Fukui 2.SEIREN CO.LTD Nitta 2nd Plant 60-1,1- Nittazuka, 1 Chome, Fukui #6 910-0067, Japan 7/F East, Warwick House, Taikoo #7 2018 Seamtapes Bemis China Hong Kong Place 979 King´s Road, Quarry Bay ROOM 203, TAEHWA B/D, BONTEX INTERNATIONAL JANGHAN-RO 58, DONGDAEMUN- #8 2017 Weaving, Dyeing, Coating CO., LTD Korea Seoul GU Kwai Chung, N.T. Hong 24/F., Wealty Ind. Bldg., Wing Yip #9 2017 Knit Burltex China Kong Street 2F/8 Alley 30 Lane 358 Rueiguang #10 2018 metal parts, plastic parts Button International Taiwan Neihu District Taipei 11 492 Rd Dyeing; Weaving, 12FL , NO. 81 , CHENG THE ROAD #11 2017 Lamination Carol Textile CO., LTD Taiwan Taipei ., SEC 2 #12 2017 Knitting, Dyeing Carvico S.p.A.