Clinical Reasoning: a 22-Year-Old Man Presenting with Headache and Right Leg Jerks
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Status Epilepticus Clinical Pathway
JOHNS HOPKINS ALL CHILDREN’S HOSPITAL Status Epilepticus Clinical Pathway 1 Johns Hopkins All Children's Hospital Status Epilepticus Clinical Pathway Table of Contents 1. Rationale 2. Background 3. Diagnosis 4. Labs 5. Radiologic Studies 6. General Management 7. Status Epilepticus Pathway 8. Pharmacologic Management 9. Therapeutic Drug Monitoring 10. Inpatient Status Admission Criteria a. Admission Pathway 11. Outcome Measures 12. References Last updated: July 7, 2019 Owners: Danielle Hirsch, MD, Emergency Medicine; Jennifer Avallone, DO, Neurology This pathway is intended as a guide for physicians, physician assistants, nurse practitioners and other healthcare providers. It should be adapted to the care of specific patient based on the patient’s individualized circumstances and the practitioner’s professional judgment. 2 Johns Hopkins All Children's Hospital Status Epilepticus Clinical Pathway Rationale This clinical pathway was developed by a consensus group of JHACH neurologists/epileptologists, emergency physicians, advanced practice providers, hospitalists, intensivists, nurses, and pharmacists to standardize the management of children treated for status epilepticus. The following clinical issues are addressed: ● When to evaluate for status epilepticus ● When to consider admission for further evaluation and treatment of status epilepticus ● When to consult Neurology, Hospitalists, or Critical Care Team for further management of status epilepticus ● When to obtain further neuroimaging for status epilepticus ● What ongoing therapy patients should receive for status epilepticus Background: Status epilepticus (SE) is the most common neurological emergency in children1 and has the potential to cause substantial morbidity and mortality. Incidence among children ranges from 17 to 23 per 100,000 annually.2 Prevalence is highest in pediatric patients from zero to four years of age.3 Ng3 acknowledges the most current definition of SE as a continuous seizure lasting more than five minutes or two or more distinct seizures without regaining awareness in between. -

Cognitive Impairment: Causes, Diagnosis and Treatment
NEUROLOGY - LABORATORY AND CLINICAL RESEARCH DEVELOPMENTS COGNITIVE IMPAIRMENT: CAUSES, DIAGNOSIS AND TREATMENT No part of this digital document may be reproduced, stored in a retrieval system or transmitted in any form or by any means. The publisher has taken reasonable care in the preparation of this digital document, but makes no expressed or implied warranty of any kind and assumes no responsibility for any errors or omissions. No liability is assumed for incidental or consequential damages in connection with or arising out of information contained herein. This digital document is sold with the clear understanding that the publisher is not engaged in rendering legal, medical or any other professional services. NEUROLOGY – LABORATORY AND CLINICAL RESEARCH DEVELOPMENTS SERIES Intracranial Hypertension Stefan Mircea Iencean and Alexandru Vladimir Ciurea 2009 ISBN: 978-1-60741-862-7 (Hardcover Book) Intracranial Hypertension Stefan Mircea Iencean and Alexandru Vladimir Ciurea 2009 ISBN: 978-1-60876-549-2 (Online Book) Cerebral Blood Flow Regulation Nodar P. Mitagvaria and Haim (James) I. Bicher 2009 ISBN: 978-1-60692-163-0 Cerebral Ischemia in Young Adults: Pathogenic and Clinical Perspectives Alessandro Pezzini and Alessandro Padovani (Editors) 2009 ISBN: 978-1-60741-627-2 Dizziness: Vertigo, Disequilibrium and Lightheadedness Agnes Lindqvist and Gjord Nyman (Editors) 2009: ISBN 978-1-60741-847-4 Somatosensory Cortex: Roles, Interventions and Traumas Niels Johnsen and Rolf Agerskov (Editors) 2009. ISBN: 978-1-60741-876-4 Cognitive Impairment: Causes, Diagnosis and Treatment Melanie L. Landow (Editor) 2009. ISBN: 978-1-60876-205-7 NEUROLOGY - LABORATORY AND CLINICAL RESEARCH DEVELOPMENTS COGNITIVE IMPAIRMENT: CAUSES, DIAGNOSIS AND TREATMENT MELANIE L. -

Spinal Segmental Myoclonus Masquerading As a Psychogenic Movement Disorder Laxmi Khanna1*, Anuradha Batra1 and Ankita Sharma2
Short Communication iMedPub Journals Journal of Neurology and Neuroscience 2019 www.imedpub.com Vol.10 No.1:282 ISSN 2171-6625 DOI: 10.21767/2171-6625.1000282 Spinal Segmental Myoclonus Masquerading as a Psychogenic Movement Disorder Laxmi Khanna1*, Anuradha Batra1 and Ankita Sharma2 1Department of Neurology and Neurophysiology, Sir Gangaram Hospital, New Delhi, India 2Department of Neurophysiology, Sir Gangaram Hospital, New Delhi, India *Corresponding author: Dr. Laxmi Khanna, Consultant Neurologist and Neurophysiologist, Department of Neurology and Neurophysiology, Sir Gangaram Hospital, New Delhi–110060, India, Tel: 9873558121; E-mail: [email protected] Rec Date: December 26, 2018; Acc Date: January 05, 2019; Pub Date: January 11, 2019 Citation: Khanna L, Batra A, Sharma A (2019) Spinal Segmental Myoclonus Masquerading as a Psychogenic Movement Disorder. J Neurol Neurosci Vol.10 No.1:282. his lower back followed by a painless involuntary rhythmic forward jerking of his legs which lasted for thirty seconds Abstract (Figures 1 and 2). These attacks occurred at rest and before falling asleep. The jerks increased in frequency and intensity Spinal generated movement disorders are uncommon. An curtailing his social life. There was no past history of any elderly gentleman presented with distressing jerks of both trauma, myelitis, demyelination or infections with human lower limbs which caused him much social immunodeficiency virus. embarrassment. He had received psychiatric treatment for these abnormal muscular spasms without relief. He had become depressed and withdrawn when he first presented to our outpatient department. A routine clinical examination followed by a long-term video EEG with simultaneous EMG clinched the diagnosis of a spinal segmental myoclonus. -

Clinical Controversies in Amyotrophic Lateral Sclerosis
Clinical Controversies in Amyotrophic Lateral Sclerosis Authors: Ruaridh Cameron Smail,1,2 Neil Simon2 1. Department of Neurology and Neurophysiology, Royal North Shore Hospital, Sydney, Australia 2. Northern Clinical School, The University of Sydney, Sydney, Australia *Correspondence to [email protected] Disclosure: The authors have declared no conflicts of interest. Received: 26.02.20 Accepted: 28.04.20 Keywords: Amyotrophic lateral sclerosis (ALS), biomarker, clinical phenotype, diagnosis, motor neurone disease (MND), pathology, ultrasound. Citation: EMJ Neurol. 2020;8[1]:80-92. Abstract Amyotrophic lateral sclerosis is a devastating neurodegenerative condition with few effective treatments. Current research is gathering momentum into the underlying pathology of this condition and how components of these pathological mechanisms affect individuals differently, leading to the broad manifestations encountered in clinical practice. We are moving away from considering this condition as merely an anterior horn cell disorder into a framework of a multisystem neurodegenerative condition in which early cortical hyperexcitability is key. The deposition of TAR DNA-binding protein 43 is also a relevant finding given the overlap with frontotemporal dysfunction. New techniques have been developed to provide a more accurate diagnosis, earlier in the disease course. This goes beyond the traditional nerve conduction studies and needle electromyography, to cortical excitability studies using transcranial magnetic stimulation, and the use of ultrasound. These ancillary tests are proposed for consideration of future diagnostic paradigms. As we learn more about this disease, future treatments need to ensure efficacy, safety, and a suitable target population to improve outcomes for these patients. In this time of active research into this condition, this paper highlights some of the areas of controversy to induce discussion surrounding these topics. -

Myoclonus Aspen Summer 2020
Hallett Myoclonus Aspen Summer 2020 Myoclonus (Chapter 20) Aspen 2020 1 Myoclonus: Definition Quick muscle jerks Either irregular or rhythmic, but always simple 2 1 Hallett Myoclonus Aspen Summer 2020 Myoclonus • Spontaneous • Action myoclonus: activated or accentuated by voluntary movement • Reflex myoclonus: activated or accentuated by sensory stimulation 3 Myoclonus • Focal: involving only few adjacent muscles • Generalized: involving most or many of the muscles of the body • Multifocal: involving many muscles, but in different jerks 4 2 Hallett Myoclonus Aspen Summer 2020 Differential diagnosis of myoclonus • Simple tics • Some components of chorea • Tremor • Peripheral disorders – Fasciculation – Myokymia – Hemifacial spasm 9 Classification of Myoclonus Site of Origin • Cortex – Cortical myoclonus, epilepsia partialis continua, cortical tremor • Brainstem – Reticular myoclonus, exaggerated startle, palatal myoclonus • Spinal cord – Segmental, propriospinal • Peripheral – Rare, likely due to secondary CNS changes 10 3 Hallett Myoclonus Aspen Summer 2020 Classification of myoclonus to guide therapy • First consideration: Etiological classification – Is there a metabolic encephalopathy to be treated? Is there a tumor to be removed? Is a drug responsible? • Second consideration: Physiological classification – Can the myoclonus be treated symptomatically even if the underlying condition remains unchanged? 12 Myoclonus: Physiological Classification • Epileptic • Non‐epileptic The basic question to ask is whether the myoclonus is a “fragment -

A Rare Case of Immune Reconstitution Inflammatory Syndrome Development in an Immunocompromised Patient with Progressive Multifocal Leukoencephalopathy And
Hindawi Publishing Corporation Case Reports in Neurological Medicine Volume 2013, Article ID 460701, 5 pages http://dx.doi.org/10.1155/2013/460701 Case Report A Rare Case of Immune Reconstitution Inflammatory Syndrome Development in an Immunocompromised Patient with Progressive Multifocal Leukoencephalopathy and Multicentric Castleman’s Disease Mohankumar Kurukumbi,1 Sheldon Steiner,1,2 Shariff Dunlap,1 Sherita Chapman,3 Noha Solieman,1 and Annapurni Jayam-Trouth1 1 Department of Neurology, Howard University Hospital, 2041 Georgia Avenue, Washington, DC 20060, USA 2 Department of Neurology, Howard University College of Medicine, Washington, DC 20060, USA 3 Department of Neurology, University of Virginia, Charlottesville, VA 22908, USA Correspondence should be addressed to Mohankumar Kurukumbi; [email protected] Received 27 June 2013; Accepted 17 July 2013 AcademicEditors:T.K.Banerjee,J.L.Gonzalez-Guti´ errez,´ R. Hashimoto, J. C. Kattah, and Y. Wakabayashi Copyright © 2013 Mohankumar Kurukumbi et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Immune reconstitution inflammatory syndrome (IRIS) development in HIV with preexistent progressive multifocal leukoen- cephalopathy (PML) has been extensively studied. PML-IRIS typically manifests clinically as new or worsening neurologic symptoms in conjunction with enlarging CNS lesions and occurs in approximately 10–20 percent of HIV-infected patients with PML who begin HAART. Likewise, Multicentric Castleman’s Disease (MCD), a rare malignant lymphoproliferative disorder, has a strong and well-known association with HIV. Our case provides a rare instance of PML-IRIS in combination with MCD in an HIV-positive individual. -

Epilepsy Syndromes E9 (1)
EPILEPSY SYNDROMES E9 (1) Epilepsy Syndromes Last updated: September 9, 2021 CLASSIFICATION .......................................................................................................................................... 2 LOCALIZATION-RELATED (FOCAL) EPILEPSY SYNDROMES ........................................................................ 3 TEMPORAL LOBE EPILEPSY (TLE) ............................................................................................................... 3 Epidemiology ......................................................................................................................................... 3 Etiology, Pathology ................................................................................................................................ 3 Clinical Features ..................................................................................................................................... 7 Diagnosis ................................................................................................................................................ 8 Treatment ............................................................................................................................................. 15 EXTRATEMPORAL NEOCORTICAL EPILEPSY ............................................................................................... 16 Etiology ................................................................................................................................................ 16 -
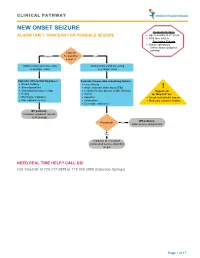
Seizure, New Onset
CLINICAL PATHWAY NEW ONSET SEIZURE Inclusion Criteria ALGORITHM 1. CONCERN FOR POSSIBLE SEIZURE • Age 6 months to 21 years • First-time seizure Exclusion Criteria • Status epilepticus (refer to status epilepticus pathway) Concern Unsure for possible Yes seizure? Obtain history and screening Obtain history and screening neurologic exam neurologic exam Consider differential diagnoses: Evaluate for possible provoking factors: • Breath holding • Fever/illness • Stereotypies/tics • Acute traumatic brain injury (TBI) ! • Vasovagal/syncope/vertigo • Central nervous system (CNS) infection Urgent call • Reflux • Tumor to ‘OneCall’ for: • Electrolyte imbalance • Ingestion • Suspected infantile spasm • Non epileptic seizure • Intoxication • Medically complex children • Electrolyte imbalance Off pathway: Consider outpatient referral to Neurology Off pathway: Provoked? Yes Address provoking factors No Continue to new onset unprovoked seizure algorithm on p.2 NEED REAL TIME HELP? CALL US! Call ‘OneCall’ at 720-777-3999 or 719-305-3999 (Colorado Springs) Page 1 of 17 CLINICAL PATHWAY ALGORITHM 2. NEW ONSET UNPROVOKED SEIZURE Inclusion Criteria • Age 6 months to 21 years New onset unprovoked • First-time unprovoked seizure seizure • Newly recognized seizure or epilepsy syndrome Exclusion Criteria • Provoked seizure: any seizure as a symptom of fever/illness, acute traumatic brain injury (TBI), Has central nervous system (CNS) infection, tumor, patient returned ingestion, intoxication, or electrolyte imbalance Consult inpatient • to baseline within No -

ASAH1 Variant Causing a Mild SMA Phenotype with No Myoclonic Epilepsy: a Clinical, Biochemical and Molecular Study
European Journal of Human Genetics (2016) 24, 1578–1583 & 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 1018-4813/16 www.nature.com/ejhg ARTICLE ASAH1 variant causing a mild SMA phenotype with no myoclonic epilepsy: a clinical, biochemical and molecular study Massimiliano Filosto*,1, Massimo Aureli2, Barbara Castellotti3, Fabrizio Rinaldi1, Domitilla Schiumarini2, Manuela Valsecchi2, Susanna Lualdi4, Raffaella Mazzotti4, Viviana Pensato3, Silvia Rota1, Cinzia Gellera3, Mirella Filocamo4 and Alessandro Padovani1 ASAH1 gene encodes for acid ceramidase that is involved in the degradation of ceramide into sphingosine and free fatty acids within lysosomes. ASAH1 variants cause both the severe and early-onset Farber disease and rare cases of spinal muscular atrophy (SMA) with progressive myoclonic epilepsy (SMA-PME), phenotypically characterized by childhood onset of proximal muscle weakness and atrophy due to spinal motor neuron degeneration followed by occurrence of severe and intractable myoclonic seizures and death in the teenage years. We studied two subjects, a 30-year-old pregnant woman and her 17-year-old sister, affected with a very slowly progressive non-5q SMA since childhood. No history of seizures or myoclonus has been reported and EEG was unremarkable. The molecular study of ASAH1 gene showed the presence of the homozygote nucleotide variation c.124A4G (r.124a4g) that causes the amino acid substitution p.Thr42Ala. Biochemical evaluation of cultured fibroblasts showed both reduction in ceramidase activity and accumulation of ceramide compared with the normal control. This study describes for the first time the association between ASAH1 variants and an adult SMA phenotype with no myoclonic epilepsy nor death in early age, thus expanding the phenotypic spectrum of ASAH1-related SMA. -

Restless Legs Syndrome and Periodic Limb Movements of Sleep
Volume I, Issue 2 Restless Legs Syndrome and Periodic Limb Movements of Sleep Overview Periodic limb movement disorder (PLMD) ; May cause involuntary and restless leg syndrome (RLS) are jerking of the limbs distinct disorders, but often occur during sleep and simultaneously. Both PLMD and RLS are sometimes during also called (nocturnal) myoclonus, which wakefulness describes frequent or involuntary muscle spasms. Periodic limb movement was If you do have restless legs formally described first in the 1950s, and, syndrome (RLS), you are not alone. Up to Occupy your mind. Keeping your by the 1970s, it was listed as a potential 8% of the US population may have this mind actively engaged may lessen cause of insomnia. In addition to producing neurologic condition. Many people have a your symptoms of RLS. Find an similar symptoms, PLMD and RLS are mild form of the disorder, but RLS severely activity that you enjoy to help you treated similarly. affects the lives of millions of individuals. through those times when your symptoms are particularly What is Restless Legs Syndrome (RLS)? Living with RLS involves developing troublesome. Restless Legs Syndrome is an coping strategies that work for you. Here overwhelming urge to move the legs are some of our favorites. Rise to new levels. You may be more usually caused by uncomfortable or comfortable if you elevate your unpleasant sensations in the legs. It may Talk about RLS. Sharing information desktop or bookstand to a height that appear as a creepy crawly type of sensation about RLS will help your family will allow you to stand while you work or a tingling sensation. -

Part Ii – Neurological Disorders
Part ii – Neurological Disorders CHAPTER 14 MOVEMENT DISORDERS AND MOTOR NEURONE DISEASE Dr William P. Howlett 2012 Kilimanjaro Christian Medical Centre, Moshi, Kilimanjaro, Tanzania BRIC 2012 University of Bergen PO Box 7800 NO-5020 Bergen Norway NEUROLOGY IN AFRICA William Howlett Illustrations: Ellinor Moldeklev Hoff, Department of Photos and Drawings, UiB Cover: Tor Vegard Tobiassen Layout: Christian Bakke, Division of Communication, University of Bergen E JØM RKE IL T M 2 Printed by Bodoni, Bergen, Norway 4 9 1 9 6 Trykksak Copyright © 2012 William Howlett NEUROLOGY IN AFRICA is freely available to download at Bergen Open Research Archive (https://bora.uib.no) www.uib.no/cih/en/resources/neurology-in-africa ISBN 978-82-7453-085-0 Notice/Disclaimer This publication is intended to give accurate information with regard to the subject matter covered. However medical knowledge is constantly changing and information may alter. It is the responsibility of the practitioner to determine the best treatment for the patient and readers are therefore obliged to check and verify information contained within the book. This recommendation is most important with regard to drugs used, their dose, route and duration of administration, indications and contraindications and side effects. The author and the publisher waive any and all liability for damages, injury or death to persons or property incurred, directly or indirectly by this publication. CONTENTS MOVEMENT DISORDERS AND MOTOR NEURONE DISEASE 329 PARKINSON’S DISEASE (PD) � � � � � � � � � � � -

Epilepsy After Stroke
Stroke Helpline: 0303 3033 100 Website: stroke.org.uk Epilepsy after stroke In the first few weeks after a stroke some people have a seizure, and a small number go on to develop epilepsy – a tendency to have repeated seizures. These can usually be completely controlled with treatment. This factsheet explains what epilepsy is, the different types of seizures, and how epilepsy is diagnosed and treated. It also includes advice about coping with a seizure, and a glossary. Epilepsy is a tendency to have repeated What causes seizures? seizures – sometimes called ‘fits’ or ‘attacks’. It affects just under one per cent Cells in the brain communicate with one of people in the UK. Stroke is one of many another and with our muscles by passing conditions that can lead to epilepsy. electrical signals along nerve fibres. If you have epilepsy this electrical activity can Around five per cent of people who have a become disordered. A sudden abnormal stroke will have a seizure within the following burst of electrical activity in the brain can few weeks. These are known as acute or lead to a seizure. onset seizures and normally happen within 24 hours of the stroke. You are more likely There are over 40 different types of seizures to have one if you have had a severe stroke, ranging from tingling sensations or ‘going a stroke caused by bleeding in the brain (a blank’ for a few seconds, to shaking and haemorrhagic stroke), or a stroke involving losing consciousness. the part of the brain called the cerebral cortex. If you have an onset seizure, it This can mean that epilepsy is sometimes does not necessarily mean you have or will confused with other conditions, including develop epilepsy.