157800V1.Full.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

Cytogenetic Analysis of a Pseudoangiomatous Pleomorphic/Spindle Cell Lipoma
ANTICANCER RESEARCH 37 : 2219-2223 (2017) doi:10.21873/anticanres.11557 Cytogenetic Analysis of a Pseudoangiomatous Pleomorphic/Spindle Cell Lipoma IOANNIS PANAGOPOULOS 1, LUDMILA GORUNOVA 1, INGVILD LOBMAIER 2, HEGE KILEN ANDERSEN 1, BODIL BJERKEHAGEN 2 and SVERRE HEIM 1,3 1Section for Cancer Cytogenetics, Institute for Cancer Genetics and Informatics, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway; 2Department of Pathology, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway; 3Faculty of Medicine, University of Oslo, Oslo, Norway Abstract. Background: Pseudoangiomatous pleomorphic/ appearance’ (1). To date, only 20 patients have been described spindle cell lipoma is a rare subtype of pleomorphic/spindle in the literature with this diagnosis, 15 of whom were males cell lipoma. Only approximately 20 such tumors have been (1-10). The pseudoangiomatous pleomorphic/spindle cell described. Genetic information on pseudoangiomatous lipomas were mostly found in the neck (seven patients) and pleomorphic/spindle cell lipoma is restricted to a single case shoulders (four patients), but have also been seen in the in which deletion of the forkhead box O1 (FOXO1) gene was cheek, chest, chin, elbow, finger, subscapular region, and found, using fluorescence in situ hybridization (FISH). thumb. Genetic information on pseudoangiomatous Materials and Methods: G-banding and FISH analyses were pleomorphic/ spindle cell lipoma is restricted to one case only performed on a pseudoangiomatous pleomorphic/spindle cell (8) in which fluorescence in situ hybridization (FISH) with a lipoma. Results: G-banding of tumor cells showed complex probe for the forkhead box O1 ( FOXO1 ) gene, which maps karyotypic changes including loss of chromosome 13. FISH to chromosome sub-band 13q14.11, showed a signal pattern analysis revealed that the deleted region contained the RB1 indicating monoallelic loss of the gene in 57% of the gene (13q14.2) and the part of chromosome arm 13q (q14.2- examined cells. -

Downloaded from Here
bioRxiv preprint doi: https://doi.org/10.1101/017566; this version posted November 19, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Testing for ancient selection using cross-population allele 2 frequency differentiation 1;∗ 3 Fernando Racimo 4 1 Department of Integrative Biology, University of California, Berkeley, CA, USA 5 ∗ E-mail: [email protected] 6 1 Abstract 7 A powerful way to detect selection in a population is by modeling local allele frequency changes in a 8 particular region of the genome under scenarios of selection and neutrality, and finding which model is 9 most compatible with the data. Chen et al. [2010] developed a composite likelihood method called XP- 10 CLR that uses an outgroup population to detect departures from neutrality which could be compatible 11 with hard or soft sweeps, at linked sites near a beneficial allele. However, this method is most sensitive 12 to recent selection and may miss selective events that happened a long time ago. To overcome this, 13 we developed an extension of XP-CLR that jointly models the behavior of a selected allele in a three- 14 population tree. Our method - called 3P-CLR - outperforms XP-CLR when testing for selection that 15 occurred before two populations split from each other, and can distinguish between those events and 16 events that occurred specifically in each of the populations after the split. -

Genotype-Phenotype Correlations in Patients with Retinoblastoma And
Genotype-phenotype correlations in patients with Retinoblastoma and an interstitial 13q deletion Diana Mitter, Reinhard Ullmann, Artur Muradyan, Ludger Klein-Hitpaß, Deniz Kanber, Dietmar R Lohmann, Katrin Ounap, Marc Kaulisch To cite this version: Diana Mitter, Reinhard Ullmann, Artur Muradyan, Ludger Klein-Hitpaß, Deniz Kanber, et al.. Genotype-phenotype correlations in patients with Retinoblastoma and an interstitial 13q deletion. European Journal of Human Genetics, Nature Publishing Group, 2011, 10.1038/ejhg.2011.58. hal- 00633984 HAL Id: hal-00633984 https://hal.archives-ouvertes.fr/hal-00633984 Submitted on 20 Oct 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 Genotype-phenotype correlations in patients with Retinoblastoma and interstitial 13q deletions 1, *Diana Mitter, 2Reinhard Ullmann, 2Artur Muradyan, 3Ludger Klein-Hitpaß, 1Deniz Kanber, 4Katrin Õunap, 5Marc Kaulisch, 1Dietmar Lohmann 1Institut für Humangenetik, Universitätsklinikum Essen, Germany; 2Max-Planck- Institute of Molecular Genetic, Berlin, Germany; 3Institute for Cell Biology (Tumor Research), University of Duisburg-Essen, Germany; 4Department of Genetics, United Laboratories, Tartu University Hospital, Estonia; 5Institute for Research Information and Quality Assurance, Germany. *Correspondence and present address: Diana Mitter, Institut für Humangenetik, Universitätsklinikum Leipzig, Philipp-Rosenthal-Str. -

Human Social Genomics in the Multi-Ethnic Study of Atherosclerosis
Getting “Under the Skin”: Human Social Genomics in the Multi-Ethnic Study of Atherosclerosis by Kristen Monét Brown A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Epidemiological Science) in the University of Michigan 2017 Doctoral Committee: Professor Ana V. Diez-Roux, Co-Chair, Drexel University Professor Sharon R. Kardia, Co-Chair Professor Bhramar Mukherjee Assistant Professor Belinda Needham Assistant Professor Jennifer A. Smith © Kristen Monét Brown, 2017 [email protected] ORCID iD: 0000-0002-9955-0568 Dedication I dedicate this dissertation to my grandmother, Gertrude Delores Hampton. Nanny, no one wanted to see me become “Dr. Brown” more than you. I know that you are standing over the bannister of heaven smiling and beaming with pride. I love you more than my words could ever fully express. ii Acknowledgements First, I give honor to God, who is the head of my life. Truly, without Him, none of this would be possible. Countless times throughout this doctoral journey I have relied my favorite scripture, “And we know that all things work together for good, to them that love God, to them who are called according to His purpose (Romans 8:28).” Secondly, I acknowledge my parents, James and Marilyn Brown. From an early age, you two instilled in me the value of education and have been my biggest cheerleaders throughout my entire life. I thank you for your unconditional love, encouragement, sacrifices, and support. I would not be here today without you. I truly thank God that out of the all of the people in the world that He could have chosen to be my parents, that He chose the two of you. -

PDF Datasheet
Product Datasheet RCBTB2 Overexpression Lysate NBL1-15241 Unit Size: 0.1 mg Store at -80C. Avoid freeze-thaw cycles. Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NBL1-15241 Updated 3/17/2020 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NBL1-15241 Page 1 of 2 v.20.1 Updated 3/17/2020 NBL1-15241 RCBTB2 Overexpression Lysate Product Information Unit Size 0.1 mg Concentration The exact concentration of the protein of interest cannot be determined for overexpression lysates. Please contact technical support for more information. Storage Store at -80C. Avoid freeze-thaw cycles. Buffer RIPA buffer Target Molecular Weight 60.1 kDa Product Description Description Transient overexpression lysate of regulator of chromosome condensation (RCC1) and BTB (POZ) domain containing protein 2 (RCBTB2) The lysate was created in HEK293T cells, using Plasmid ID RC206560 and based on accession number NM_001268. The protein contains a C-MYC/DDK Tag. Gene ID 1102 Gene Symbol RCBTB2 Species Human Notes HEK293T cells in 10-cm dishes were transiently transfected with a non-lipid polymer transfection reagent specially designed and manufactured for large volume DNA transfection. Transfected cells were cultured for 48hrs before collection. The cells were lysed in modified RIPA buffer (25mM Tris-HCl pH7.6, 150mM NaCl, 1% NP-40, 1mM EDTA, 1xProteinase inhibitor cocktail mix, 1mM PMSF and 1mM Na3VO4, and then centrifuged to clarify the lysate. Protein concentration was measured by BCA protein assay kit.This product is manufactured by and sold under license from OriGene Technologies and its use is limited solely for research purposes. -

Oxidized Phospholipids Regulate Amino Acid Metabolism Through MTHFD2 to Facilitate Nucleotide Release in Endothelial Cells
ARTICLE DOI: 10.1038/s41467-018-04602-0 OPEN Oxidized phospholipids regulate amino acid metabolism through MTHFD2 to facilitate nucleotide release in endothelial cells Juliane Hitzel1,2, Eunjee Lee3,4, Yi Zhang 3,5,Sofia Iris Bibli2,6, Xiaogang Li7, Sven Zukunft 2,6, Beatrice Pflüger1,2, Jiong Hu2,6, Christoph Schürmann1,2, Andrea Estefania Vasconez1,2, James A. Oo1,2, Adelheid Kratzer8,9, Sandeep Kumar 10, Flávia Rezende1,2, Ivana Josipovic1,2, Dominique Thomas11, Hector Giral8,9, Yannick Schreiber12, Gerd Geisslinger11,12, Christian Fork1,2, Xia Yang13, Fragiska Sigala14, Casey E. Romanoski15, Jens Kroll7, Hanjoong Jo 10, Ulf Landmesser8,9,16, Aldons J. Lusis17, 1234567890():,; Dmitry Namgaladze18, Ingrid Fleming2,6, Matthias S. Leisegang1,2, Jun Zhu 3,4 & Ralf P. Brandes1,2 Oxidized phospholipids (oxPAPC) induce endothelial dysfunction and atherosclerosis. Here we show that oxPAPC induce a gene network regulating serine-glycine metabolism with the mitochondrial methylenetetrahydrofolate dehydrogenase/cyclohydrolase (MTHFD2) as a cau- sal regulator using integrative network modeling and Bayesian network analysis in human aortic endothelial cells. The cluster is activated in human plaque material and by atherogenic lipo- proteins isolated from plasma of patients with coronary artery disease (CAD). Single nucleotide polymorphisms (SNPs) within the MTHFD2-controlled cluster associate with CAD. The MTHFD2-controlled cluster redirects metabolism to glycine synthesis to replenish purine nucleotides. Since endothelial cells secrete purines in response to oxPAPC, the MTHFD2- controlled response maintains endothelial ATP. Accordingly, MTHFD2-dependent glycine synthesis is a prerequisite for angiogenesis. Thus, we propose that endothelial cells undergo MTHFD2-mediated reprogramming toward serine-glycine and mitochondrial one-carbon metabolism to compensate for the loss of ATP in response to oxPAPC during atherosclerosis. -
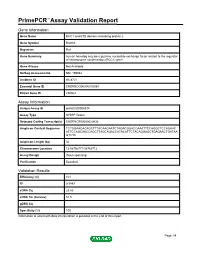
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name RCC1 and BTB domain-containing protein 2 Gene Symbol Rcbtb2 Organism Rat Gene Summary human homolog may be a guanine nucleotide exchange factor related to the regulator of chromosome condensation (RCC1) gene Gene Aliases Not Available RefSeq Accession No. NM_199084 UniGene ID Rn.8721 Ensembl Gene ID ENSRNOG00000015054 Entrez Gene ID 290363 Assay Information Unique Assay ID qRnoCID0004834 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000020836 Amplicon Context Sequence TCTGGAAGACAGCTTTACAAGAATCTAGACGGACCAAATTCCAGGCTCCAGAAC ATTCCAGCAACCAGCTTAGCAGACCATACATTCTACAGGAGCTGAGAACTGATAA GTCTC Amplicon Length (bp) 84 Chromosome Location 15:58766771-58769712 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 101 R2 0.9997 cDNA Cq 23.82 cDNA Tm (Celsius) 81.5 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Rcbtb2, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Receptor Signaling Through Osteoclast-Associated Monocyte
Downloaded from http://www.jimmunol.org/ by guest on September 29, 2021 is online at: average * The Journal of Immunology The Journal of Immunology , 20 of which you can access for free at: 2015; 194:3169-3179; Prepublished online 27 from submission to initial decision 4 weeks from acceptance to publication February 2015; doi: 10.4049/jimmunol.1402800 http://www.jimmunol.org/content/194/7/3169 Collagen Induces Maturation of Human Monocyte-Derived Dendritic Cells by Signaling through Osteoclast-Associated Receptor Heidi S. Schultz, Louise M. Nitze, Louise H. Zeuthen, Pernille Keller, Albrecht Gruhler, Jesper Pass, Jianhe Chen, Li Guo, Andrew J. Fleetwood, John A. Hamilton, Martin W. Berchtold and Svetlana Panina J Immunol cites 43 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Author Choice option Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Freely available online through http://www.jimmunol.org/content/suppl/2015/02/27/jimmunol.140280 0.DCSupplemental This article http://www.jimmunol.org/content/194/7/3169.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Author Choice Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2015 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. -

The Pdx1 Bound Swi/Snf Chromatin Remodeling Complex Regulates Pancreatic Progenitor Cell Proliferation and Mature Islet Β Cell
Page 1 of 125 Diabetes The Pdx1 bound Swi/Snf chromatin remodeling complex regulates pancreatic progenitor cell proliferation and mature islet β cell function Jason M. Spaeth1,2, Jin-Hua Liu1, Daniel Peters3, Min Guo1, Anna B. Osipovich1, Fardin Mohammadi3, Nilotpal Roy4, Anil Bhushan4, Mark A. Magnuson1, Matthias Hebrok4, Christopher V. E. Wright3, Roland Stein1,5 1 Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 2 Present address: Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN 3 Department of Cell and Developmental Biology, Vanderbilt University, Nashville, TN 4 Diabetes Center, Department of Medicine, UCSF, San Francisco, California 5 Corresponding author: [email protected]; (615)322-7026 1 Diabetes Publish Ahead of Print, published online June 14, 2019 Diabetes Page 2 of 125 Abstract Transcription factors positively and/or negatively impact gene expression by recruiting coregulatory factors, which interact through protein-protein binding. Here we demonstrate that mouse pancreas size and islet β cell function are controlled by the ATP-dependent Swi/Snf chromatin remodeling coregulatory complex that physically associates with Pdx1, a diabetes- linked transcription factor essential to pancreatic morphogenesis and adult islet-cell function and maintenance. Early embryonic deletion of just the Swi/Snf Brg1 ATPase subunit reduced multipotent pancreatic progenitor cell proliferation and resulted in pancreas hypoplasia. In contrast, removal of both Swi/Snf ATPase subunits, Brg1 and Brm, was necessary to compromise adult islet β cell activity, which included whole animal glucose intolerance, hyperglycemia and impaired insulin secretion. Notably, lineage-tracing analysis revealed Swi/Snf-deficient β cells lost the ability to produce the mRNAs for insulin and other key metabolic genes without effecting the expression of many essential islet-enriched transcription factors. -

Mouse Rb1 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Rb1 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Rb1 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Rb1 gene (NCBI Reference Sequence: NM_009029 ; Ensembl: ENSMUSG00000022105 ) is located on Mouse chromosome 14. 27 exons are identified, with the ATG start codon in exon 1 and the TGA stop codon in exon 27 (Transcript: ENSMUST00000022701). Exon 8 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Rb1 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-217E12 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Homozygotes for targeted mutations exhibit abnormalities of the neuronal and hematopoietic systems and die in utero. Heterozygotes may develop pituitary tumors associated with loss of the normal allele. Exon 8 starts from about 25.37% of the coding region. The knockout of Exon 8 will result in frameshift of the gene. The size of intron 7 for 5'-loxP site insertion: 2603 bp, and the size of intron 8 for 3'-loxP site insertion: 1630 bp. The size of effective cKO region: ~643 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 8 9 27 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Rb1 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats.