Synthesis of Α‑Glucan in Mycobacteria Involves a Hetero-Octameric
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

University of Birmingham Synthesis of -Glucan in Mycobacteria Involves A
University of Birmingham Synthesis of -glucan in mycobacteria involves a hetero-octameric complex of trehalose synthase TreS and Maltokinase Pep2 Roy, Rana; Veeraraghavan, Usha; Kermani, Ali; Scott, David J; Hyde, Eva I; Besra, Gurdyal S; Alderwick, Luke J; Fütterer, Klaus DOI: 10.1021/cb400508k License: Creative Commons: Attribution (CC BY) Document Version Publisher's PDF, also known as Version of record Citation for published version (Harvard): Roy, R, Veeraraghavan, U, Kermani, A, Scott, DJ, Hyde, EI, Besra, GS, Alderwick, LJ & Fütterer, K 2013, 'Synthesis of -glucan in mycobacteria involves a hetero-octameric complex of trehalose synthase TreS and Maltokinase Pep2', ACS chemical biology, vol. 8, no. 10, pp. 2245-2255. https://doi.org/10.1021/cb400508k Link to publication on Research at Birmingham portal Publisher Rights Statement: Eligibility for repository : checked 30/06/2014 General rights Unless a licence is specified above, all rights (including copyright and moral rights) in this document are retained by the authors and/or the copyright holders. The express permission of the copyright holder must be obtained for any use of this material other than for purposes permitted by law. •Users may freely distribute the URL that is used to identify this publication. •Users may download and/or print one copy of the publication from the University of Birmingham research portal for the purpose of private study or non-commercial research. •User may use extracts from the document in line with the concept of ‘fair dealing’ under the Copyright, Designs and Patents Act 1988 (?) •Users may not further distribute the material nor use it for the purposes of commercial gain. -

Cultural Soil
Structural and functional metage- nomic analyses of a tropical agri- cultural soil Análisis metagenómicos estructurales y funcionales de un suelo agrícola tropical Análises metagenómicas estruturais e funcionais de um solo agrícola tropical AUTHORS Received: 04.07.2018 Revised: 09.12.2018 Accepted: 21.01.2019 Lateef Babatunde 1 Salam@, 1 ABSTRACT babssalaam@yahoo. com Understanding the intricate link between the soil microbiota and their metabolic functions is important for agricultural and ecological processes and could be used as a biomarker of soil health. Oluwafemi Sunday To understand the relationship between soil microbial community structure and functions, a soil 2 Obayori microcosm designated 2S (agricultural soil) was set up. Metagenomic DNA was extracted from the soil microcosm and sequenced using Miseq Illumina next generation sequencing and analysed for their structural and functional properties. Structural analysis of the soil microcosm by MG-RAST @ Corresponding Author revealed 40 phyla, 78 classes, 157 orders, 273 families and 750 genera. Actinobacteria (54.0%) and 1Department of Biological Proteobacteria (17.5%) are the dominant phyla while Conexibacter (8.38%), Thermoleophilum (7.40%), Sciences, Al-Hikmah and Streptomyces (4.14%) are the dominant genera. Further assignment of the metagenomics using University, Ilorin, Kwara State, Nigeria. Cluster of Orthologous Groups (COG), Kyoto Encyclopedia of Genes and Genomes (KEGG), GhostKOALA, and NCBI’s CDD revealed diverse metabolic pathways utilized by the microbial 2Department of Microbiology, Lagos State community for the metabolism of carbohydrates, amino acids, lipids, biosynthesis of secondary University, Ojo, Lagos metabolites and resistance to antibiotics. Taxonomic analysis of the annotated genes also revealed State, Nigeria. the preponderance of members of Actinobacteria and Proteobacteria. -

Masterarbeit / Master's Thesis
MASTERARBEIT / MASTER’S THESIS Titel der Masterarbeit / Title of the Master‘s Thesis „Physiology, Metabolism and Biohydrogen Production of Desulfurococcus fermentans“ verfasst von / submitted by Barbara Reischl, BSc angestrebter akademischer Grad / in partial fulfilment of the requirements for the degree of Master of Science (MSc) Wien, 2016 / Vienna 2016 Studienkennzahl lt. Studienblatt / A 066 830 degree programme code as it appears on the student record sheet: Studienrichtung lt. Studienblatt / Molecular Microbiology, Microbial Ecology degree programme as it appears on and Immunobiology the student record sheet: Betreut von / Supervisor: Univ.-Prof. Dipl.-Biol. Dr. Christa Schleper Mitbetreut von / Co-Supervisor: Mag. Mag. Dr. Simon Karl-Maria Rasso Rittmann, Bakk. 2 Table of Contents 1. Introduction .......................................................................................................... 5 1.1 Role of Biohydrogen ................................................................................................ 5 1.2 Hydrogenases ......................................................................................................... 6 1.3 Characteristics of Desulfurococcus fermentans ....................................................... 7 1.4 Aims of this Study ................................................................................................... 8 2. Material and Methods ............................................................................................ 9 2.1 Chemicals .............................................................................................................. -

Glucan in Mycobacteria Involves a Hetero-Octameric Complex of Trehalose Synthase Tres and Maltokinase Pep2
Synthesis of -glucan in mycobacteria involves a hetero-octameric complex of trehalose synthase TreS and Maltokinase Pep2 Roy, Rana; Veeraraghavan, Usha; Kermani, Ali; Scott, David; Hyde, Eva; Besra, Gurdyal; Alderwick, Luke; Futterer, Klaus DOI: 10.1021/cb400508k License: Creative Commons: Attribution (CC BY) Document Version Publisher's PDF, also known as Version of record Citation for published version (Harvard): Roy, R, Veeraraghavan, U, Kermani, A, Scott, DJ, Hyde, EI, Besra, GS, Alderwick, LJ & Fütterer, K 2013, 'Synthesis of -glucan in mycobacteria involves a hetero-octameric complex of trehalose synthase TreS and Maltokinase Pep2', ACS chemical biology, vol. 8, no. 10, pp. 2245-2255. https://doi.org/10.1021/cb400508k Link to publication on Research at Birmingham portal Publisher Rights Statement: Eligibility for repository : checked 30/06/2014 General rights Unless a licence is specified above, all rights (including copyright and moral rights) in this document are retained by the authors and/or the copyright holders. The express permission of the copyright holder must be obtained for any use of this material other than for purposes permitted by law. •Users may freely distribute the URL that is used to identify this publication. •Users may download and/or print one copy of the publication from the University of Birmingham research portal for the purpose of private study or non-commercial research. •User may use extracts from the document in line with the concept of ‘fair dealing’ under the Copyright, Designs and Patents Act 1988 (?) •Users may not further distribute the material nor use it for the purposes of commercial gain. Where a licence is displayed above, please note the terms and conditions of the licence govern your use of this document. -
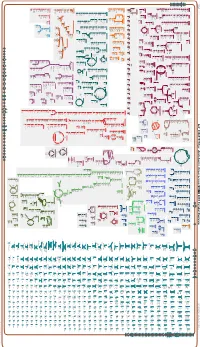
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Peter D Karp Ingrid Keseler An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Anamika Kothari Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000817975Cyc: Azotobacter chroococcum NCIMB 8003 Cellular Overview Connections between pathways are omitted for legibility. -
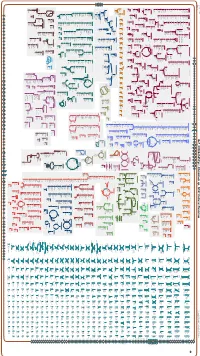
Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_001664545Cyc: Rhizobium sp. N741 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari pro a dicarboxylate an amino an amino an amino an amino an amino an amino an amino an amino glycine betaine acid acid acid molybdate an amino acid acid acid acid acid an amino sulfate arsenite sn-glycerol acid an amino an amino an amino an amino acid molybdate ammonium molybdate D-xylose predicted 3-phosphate acid acid acid acid predicted predicted predicted predicted predicted predicted predicted predicted predicted ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC transporter CcoS RS32175 RS24260 RS24235 RS24230 RS24070 RS22995 RS10550 RS21135 RS28395 RS07875 TolQ RS02515 RS03820 RS19105 YajC RS09790 ArsK RS20585 RS24115 XylF RS24250 RS24325 RS25035 UgpC RS15380 RS15005 RS27235 RS20540 RS31790 transporter transporter transporter transporter transporter transporter transporter transporter RS12545 ModC RS26725 transporter of L-proline/ predicted of an of an of an of an of an of an of an of an -

Structural and Functional Diversity of the Microbial Kinome
PLoS BIOLOGY Structural and Functional Diversity of the Microbial Kinome Natarajan Kannan1,2, Susan S. Taylor1,2, Yufeng Zhai3, J. Craig Venter4, Gerard Manning3* 1 Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, California, United States of America, 2 Howard Hughes Medical Institute, University of California San Diego, La Jolla, California, United States of America, 3 Razavi-Newman Center for Bioinformatics, Salk Institute for Biological Studies, La Jolla, California, United States of America, 4 J. Craig Venter Institute, Rockville, Maryland, United States of America The eukaryotic protein kinase (ePK) domain mediates the majority of signaling and coordination of complex events in eukaryotes. By contrast, most bacterial signaling is thought to occur through structurally unrelated histidine kinases, though some ePK-like kinases (ELKs) and small molecule kinases are known in bacteria. Our analysis of the Global Ocean Sampling (GOS) dataset reveals that ELKs are as prevalent as histidine kinases and may play an equally important role in prokaryotic behavior. By combining GOS and public databases, we show that the ePK is just one subset of a diverse superfamily of enzymes built on a common protein kinase–like (PKL) fold. We explored this huge phylogenetic and functional space to cast light on the ancient evolution of this superfamily, its mechanistic core, and the structural basis for its observed diversity. We cataloged 27,677 ePKs and 18,699 ELKs, and classified them into 20 highly distinct families whose known members suggest regulatory functions. GOS data more than tripled the count of ELK sequences and enabled the discovery of novel families and classification and analysis of all ELKs. -

Dissertation Alexander Henrich
Characterization of Maltose and Trehalose Transport in Corynebacterium glutamicum Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Universität zu Köln vorgelegt von Alexander Wolfgang Henrich aus Damscheid Köln, Februar 2011 Berichterstatter: Herr Prof. Dr. Reinhard Krämer Frau Prof. Dr. Karin Schnetz Tag der Disputation: 14. April 2011 “Observation is a passive science, experimentation an active one.” Claude Bernard Kurzzusammenfassung Kurzzusammenfassung Zur Produktion von Aminosäuren mit dem kommerziell bedeutsamen Bakterium Corynebacterium glutamicum kann aus Stärkehydrolysat gewonnene Maltose als gute und günstige Kohlenstoffquelle dienen. In der vorliegenden Arbeit wurde das Maltose/Maltodextrin Aufnahme-System MusIFGK 2-E von C. glutamicum identifiziert und charakterisiert; ein ABC Transporter mit einem K m von 1,2 ± 0,2 µM und V max von 26,2 ± 1,0 nmol/(min*mg TG). Es konnte gezeigt werden, dass das System kein Operon bildet und dass es von dem transkriptionellen Regulator RamA reguliert wird. Zusätzlich konnte eine regulatorische Verbindung zur Phosphotransferase System vermittelten Glukose Verstoffwechslung aufgezeigt werden. Weitere Analysen identifizierten das Protein MusI, welches zwar für die Aufnahme von Maltose essentiell, dessen Funktion aber noch unbekannt ist. Neben der Bedeutung als Aminosäureproduzent ist C. glutamicum ein wichtiger Modellorganismus für die Zellwand und deren Synthese innerhalb der Corynebacterineae . In diesem Zusammenhang spielt Trehalose als Komponente von Mycolaten, wichtigen Bausteinen der Zellwand von Corynebakterien and Mycobakterien, eine bedeutende Rolle. In dieser Arbeit konnte zum ersten Mal gezeigt werden, dass C. glutamicum , im Gegensatz zu bereits publizierten Daten, mit einem Trehalose Aufnahme-System ausgestattet ist. Die Aufnahme wird über den ABC Transporter TusFGK 2-E mit einem K m von 0,16 ± 0,02 µM und V max von 2,5 ± 0,1 nmol/(min*mg TG) vermittelt. -

Genomic Analysis of Secondary Metabolite Producing Actinomycetes: Acbm Is a 2-Epi-5-Epi-Valiolone 7-Kinase
Genomic Analysis of Secondary Metabolite Producing Actinomycetes: AcbM is a 2-epi-5-epi-valiolone 7-kinase A dissertation submitted to the Fachbereich Naturwissenschaften II (Chemie/Biologie) Bergische Universität – Gesamthochschule Wuppertal for the degree of Doctor of Natural Sciences (Doctor rerum naturalium) presented by MSc. Chang-Sheng Zhang Wuppertal, May 2002 ACKNOWLEDGEMENTS This work was carried out during the period between September, 1998 to May, 2002, am Lehrstuhl für Chemische Mikrobiologie, Fachbereich Chemie, der Bergischen Universität GH Wuppertal. I am sincerely grateful to: Prof. Dr. W. Piepersberg, for giving me an opportunity to be involved in these two wonderful projects; for his invaluable advice, guidance, support and encouragement throughout these years; for his insightful comments and detailed correctness of this manuscript and for his scientific ideas and vision which would benefit me forever. Prof. Dr. W. Reineke for his acceptance to co-examine this thesis and Prof. Dr. H.-J. Altenbach, Prof. Dr. G. Vogel for their kindness to be co-examiners. Dr. Udo Wehmeier, for his unwavering patience, his everlasting stimulating advice, his fruitful discussions, his enthusiastic encouragement throughout my Ph.D. program and for his critical and insightful comments on this thesis. Dr. A. Stratmann for his useful discussions and guidance in designing and performing experiments on the purification of 2-epi-5-epi-valiolone-7-phosphate. Dr. O. Block for his supply of the cyclitols for enzyme tests. Dr. R. Brückner for making the IC-MS assays. M. Podeschwa for his specialized help in analyzing the NMR data. Dr. J. Altenbuchner for his providing the deletion strains and some plasmids and for his critical reviewing of the manuscript of the gene sponge part. -

Structure of Mycobacterium Thermoresistibile Glge Defines
View metadata, citation and similar papers at core.ac.uk brought to you by CORE www.nature.com/scientificreportsprovided by Apollo OPEN Structure of Mycobacterium thermoresistibile GlgE defines novel conformational states Received: 27 July 2015 Accepted: 07 October 2015 that contribute to the catalytic Published: 30 November 2015 mechanism Vitor Mendes1,2, Michal Blaszczyk1, Ana Maranha2, Nuno Empadinhas2 & Tom L. Blundell1 GlgE, an enzyme of the pathway that converts trehalose to α-glucans, is essential for Mycobacterium tuberculosis. Inhibition of GlgE, which transfers maltose from a maltose-1-phosphate donor to α-glucan/maltooligosaccharide chain acceptor, leads to a toxic accumulation of maltose-1- phosphate that culminates in cellular death. Here we describe the first high-resolution mycobacterial GlgE structure from Mycobacterium thermoresistibile at 1.96 Å. We show that the structure resembles that of M. tuberculosis and Streptomyces coelicolor GlgEs, reported before, with each protomer in the homodimer comprising five domains. However, in M. thermoresistibile GlgE we observe several conformational states of the S domain and provide evidence that its high flexibility is important for enzyme activity. The structures here reported shed further light on the interactions between the N-terminal domains and the catalytic domains of opposing chains and how they contribute to the catalytic reaction. Importantly this work identifies a useful surrogate system to aid the development of GlgE inhibitors against opportunistic and pathogenic mycobacteria. Mycobacteria comprise a diverse group of organisms, many of which are known human pathogens. The most notorious member of this genus is undoubtedly Mycobacterium tuberculosis, the major causative agent of tuberculosis (TB), a disease that remains a leading cause of mortality worldwide, claiming an estimated 1.5 million lives in 20131. -

Structure of Mycobacterial Maltokinase, the Missing Link in the Essential Glge-Pathway
OPEN Structure of mycobacterial maltokinase, SUBJECT AREAS: the missing link in the essential STRUCTURAL BIOLOGY GLYCOBIOLOGY GlgE-pathway Joana Fraga1*, Ana Maranha2*, Vitor Mendes2, Pedro Jose´ Barbosa Pereira1, Nuno Empadinhas2 Received & Sandra Macedo-Ribeiro1 2 September 2014 Accepted 1IBMC – Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugal, 2CNC - Center for Neuroscience and Cell 2 January 2015 Biology, University of Coimbra, Coimbra, Portugal. Published 26 January 2015 A novel four-step pathway identified recently in mycobacteria channels trehalose to glycogen synthesis and is also likely involved in the biosynthesis of two other crucial polymers: intracellular methylglucose lipopolysaccharides and exposed capsular glucan. The structures of three of the intervening enzymes - GlgB, Correspondence and GlgE, and TreS - were recently reported, providing the first templates for rational drug design. Here we describe the structural characterization of the fourth enzyme of the pathway, mycobacterial maltokinase requests for materials (Mak), uncovering a eukaryotic-like kinase (ELK) fold, similar to methylthioribose kinases and should be addressed to aminoglycoside phosphotransferases. The 1.15 A˚ structure of Mak in complex with a non-hydrolysable ATP N.E. (numenius@cnc. analog reveals subtle structural rearrangements upon nucleotide binding in the cleft between the N- and the uc.pt) or S.M.-R. C-terminal lobes. Remarkably, this new family of ELKs has a novel N-terminal domain topologically ([email protected]) resembling the cystatin family of protease inhibitors. By interfacing with and restraining the mobility of the phosphate-binding region of the N-terminal lobe, Mak’s unusual N-terminal domain might regulate its phosphotransfer activity and represents the most likely anchoring point for TreS, the upstream enzyme in * These authors the pathway. -

2020.06.02.131169V1.Full.Pdf
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.02.131169; this version posted June 4, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Substitution of nucleotide-sugar by trehalose-dependent 2 glycogen synthesis pathways in Chlamydiales underlines an 3 unusual requirement for storage polysaccharides within 4 obligate intracellular bacteria. 5 6 Matthieu Colpaert1, Derifa Kadouche1b, Mathieu Ducatez1b, Trestan Pillonel2, Carole Kebbi- 7 Beghdadi2, Ugo Cenci1, Binquan Huang1c, Malika Chabi1, Emmanuel Maes1, Bernadette 8 Coddeville1, Loïc Couderc3, Hélène Touzet4, Fabrice Bray5, Catherine Tirtiaux1, Steven Ball1, 9 Gilbert Greub2, Colleoni Christophe1a. 10 11 1: University of Lille, CNRS, UMR8576-UGSF-Unité de Glycobiologie Structurale et 12 Fonctionnelle, F-59000 Lille, France 13 2: Institute of Microbiology, University of Lausanne and University Hospital Center, Lausanne, 14 Switzerland 15 3: University of Lille, CNRS, Inserm, CHU Lille, Institut Pasteur de Lille, US 41 - UMS 2014 - 16 PLBS, F-59000 Lille, France 17 4: University of Lille, CNRS, Centrale Lille, UMR 9189 - CRIStAL - Centre de Recherche en 18 Informatique Signal et Automatique de Lille, F-59000 Lille, France 19 5: University of Lille, CNRS, USR 3290—MSAP—Miniaturisation pour la Synthèse, l’Analyse et 20 la Protéomique, F-59000 Lille, France 21 22 a Corresponding author : Christophe Colleoni, University of Lille, CNRS, UMR8576-UGSF-Unité 23 de Glycobiologie Structurale et Fonctionnelle, F-59000 Lille, France Bâtiment C9, Cité 24 Scientifique, 59655 Villeneuve d’Ascq Cedex .