Properties of Hydrocarbon Mixtures As Related to Production Problems
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![C9-14 Aliphatic [2-25% Aromatic] Hydrocarbon Solvents Category SIAP](https://docslib.b-cdn.net/cover/2852/c9-14-aliphatic-2-25-aromatic-hydrocarbon-solvents-category-siap-12852.webp)
C9-14 Aliphatic [2-25% Aromatic] Hydrocarbon Solvents Category SIAP
CoCAM 2, 17-19 April 2012 BIAC/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical C -C Aliphatic [2-25% aromatic] Hydrocarbon Solvents Category Category 9 14 Substance Name CAS Number Stoddard solvent 8052-41-3 Chemical Names Kerosine, petroleum, hydrodesulfurized 64742-81-0 and CAS Naphtha, petroleum, hydrodesulfurized heavy 64742-82-1 Registry Solvent naphtha, petroleum, medium aliphatic 64742-88-7 Numbers Note: Substances in this category are also commonly known as mineral spirits, white spirits, or Stoddard solvent. CAS Number Chemical Description † 8052-41-3 Includes C8 to C14 branched, linear, and cyclic paraffins and aromatics (6 to 18%), <50ppmV benzene † 64742-81-0 Includes C9 to C14 branched, linear, and cyclic paraffins and aromatics (10 to Structural 25%), <100 ppmV benzene Formula † and CAS 64742-82-1 Includes C8 to C13 branched, linear, and cyclic paraffins and aromatics (15 to 25%), <100 ppmV benzene Registry † Numbers 64742-88-7 Includes C8 to C13 branched, linear, and cyclic paraffins and aromatics (14 to 20%), <50 ppmV benzene Individual category member substances are comprised of aliphatic hydrocarbon molecules whose carbon numbers range between C9 and C14; approximately 80% of the aliphatic constituents for a given substance fall within the C9-C14 carbon range and <100 ppmV benzene. In some instances, the carbon range of a test substance is more precisely defined in the test protocol. In these instances, the specific carbon range (e.g. C8-C10, C9-C10, etc.) will be specified in the SIAP. * It should be noted that other substances defined by the same CAS RNs may have boiling ranges outside the range of 143-254° C and that these substances are not covered by the category. -

Phase Equilibria of Supercritical Carbon Dioxide and Hydrocarbon Mixtures
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1992 Phase Equilibria of Supercritical Carbon Dioxide and Hydrocarbon Mixtures. Hyo-guk Lee Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Lee, Hyo-guk, "Phase Equilibria of Supercritical Carbon Dioxide and Hydrocarbon Mixtures." (1992). LSU Historical Dissertations and Theses. 5325. https://digitalcommons.lsu.edu/gradschool_disstheses/5325 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely afreet reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. -

BUBBLE FORMATION in SUPERSATURATED HYDROCARBON MIXTURES Petroleum Reservoirs, the Mineral and Water Surfaces with for Each Crystal Averaging About 4.5 Sq Cm
T.P. 3441 BUBBLE FORMATION IN SUPERSATURATED HYDRO CARBON MIXTURES HARVEY T. KENNEDY, A AND M COlLEGE OF TEXAS, COlLEGE STATION, TEX., MEMBER AIME, AND CHARLES R. OLSON, OHIO OIL CO., SHREVEPORT, LA., JUNIOR MEMBER AIME Downloaded from http://onepetro.org/jpt/article-pdf/4/11/271/2238408/spe-232-g.pdf by guest on 29 September 2021 ABSTRACT tude, may be calculated for any rate of pressure decline imposed on the reservoir by production. The bearing of the In many investigations of the performance of petroleum res number and distribution of bubbles on reservoir performance ervoirs the assumption is made that the liquid, if below its is discussed. bubble-point pressure, is at all times in equilibrium with gas_ On the other hand, observations by numerous investigators have indicated that gas-liquid systems including hydrocarbon systems, may exhibit supersaturation to the extent of many INTRODUCTION hundred psi in the laboratory. Up to the present, there has been no reliable data on which to judge the actual extent of A liquid system is supersaturated with gas when the amount supersaturation under conditions approaching those existing of gas dissolved exceeds that corresponding to equilibrium at in petroleum reservoirs. the existing pressure and temperature. The degree of super The work reported here deals with observations and meas saturation may be conveniently expressed as the difference urements on mixtures of methane and kerosene in the presence between the bubble-point of the mixture and the prevailing of silica and calcite crystals. Bubbles were observed to form pressure. Thus, if a mixture having a bubble-point of 1,000 on crystal-hydrocarbon surfaces in preference to the glass psi at a given temperature exists in single liquid phase at hydrocarbon interface or to the body of the liquid. -

Stoddard Solvent 105 6. Analytical Methods 6.2
STODDARD SOLVENT 105 6. ANALYTICAL METHODS 6.2 ENVIRONMENTAL SAMPLES As with biological materials, detection of Stoddard solvent in environmental samples is based on the detection of component hydrocarbons. See Table 6-2 for a summary of the analytical methods used to determine Stoddard solvent hydrocarbons in environmental samples. The primary method for detecting volatile components of Stoddard solvent in air is GC using a flame ionization detector (FID) (NIOSH 1984; Otson et al. 1983). Stoddard solvent in air may be determined by absorption to an appropriate column such as charcoal, desorption in a solvent (carbon disulfide is recommended), and subsequent quantification. Although the precision of this method is good (greater than 10% relative standard deviation when the recovery is greater than 80%), in general, recovery tends to be rather poor (18-80%) because of the slow volatilization of Stoddard solvent (Otson et al. 1983). No analytical methods specific for Stoddard solvent in water or soil samples were located; however, determination of Stoddard solvent may be assumed to be similar to the detection of comparable hydrocarbon mixtures. Detection of Stoddard solvent in water is dependent on the identification and quantification of the specific hydrocarbon components of the solvent. The primary method, GC either alone or in combination with MS, may be used for the identification of the major hydrocarbon components, i.e., n-alkanes, branched alkanes, cycloalkanes, and alkylbenzenes. Separation of the aliphatic and aromatic fractions may be achieved by liquid-solid column chromatography followed by dilution of the eluates with carbon disulfide. Aqueous samples may be extracted with trichlorotrifluoroethane, while solid samples may be extracted by Soxhlet extraction or sonication methods (Air Force 1989). -

Industrial Hydrocarbon Processes
Handbook of INDUSTRIAL HYDROCARBON PROCESSES JAMES G. SPEIGHT PhD, DSc AMSTERDAM • BOSTON • HEIDELBERG • LONDON NEW YORK • OXFORD • PARIS • SAN DIEGO SAN FRANCISCO • SINGAPORE • SYDNEY • TOKYO Gulf Professional Publishing is an imprint of Elsevier Gulf Professional Publishing is an imprint of Elsevier The Boulevard, Langford Lane, Kidlington, Oxford OX5 1GB, UK 30 Corporate Drive, Suite 400, Burlington, MA 01803, USA First edition 2011 Copyright Ó 2011 Elsevier Inc. All rights reserved No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means electronic, mechanical, photocopying, recording or otherwise without the prior written permission of the publisher Permissions may be sought directly from Elsevier’s Science & Technology Rights Department in Oxford, UK: phone (+44) (0) 1865 843830; fax (+44) (0) 1865 853333; email: [email protected]. Alternatively you can submit your request online by visiting the Elsevier web site at http://elsevier.com/locate/ permissions, and selecting Obtaining permission to use Elsevier material Notice No responsibility is assumed by the publisher for any injury and/or damage to persons or property as a matter of products liability, negligence or otherwise, or from any use or operation of any methods, products, instructions or ideas contained in the material herein. Because of rapid advances in the medical sciences, in particular, independent verification of diagnoses and drug dosages should be made British Library Cataloguing in Publication Data -

Effect of Water Saturation on H2 and CO Solubility in Hydrocarbons
chemengineering Brief Report Effect of Water Saturation on H2 and CO Solubility in Hydrocarbons Omar M. Basha Department of Chemical, Biochemical and Bio Engineering, North Carolina A&T State University, Greensboro, NC 27411, USA; [email protected] Received: 31 August 2018; Accepted: 28 January 2019; Published: 22 February 2019 Abstract: The effect of water on the solubility of syngas in hydrocarbons has typically been ignored when developing models for Fischer-Tropsch slurry bubble column reactors (SBCR), despite water being a major by-product. Therefore, a generalized correlation was developed to predict water solubility in hydrocarbons at high temperatures, and was used to calculate the effect of water saturation on H2 and CO solubility in hydrocarbons using the Span Wagner equation of state. The presence of water was shown to have a much more significant effect on H2 solubility in hydrocarbons, compared to CO. Keywords: solubility; water; hydrocarbons; syngas; Fischer-Tropsch; water-hydrocarbon mixtures 1. Introduction The effect of the presence of water on syngas solubility in Fischer-Tropsch (F-T) Slurry Bubble Column Reactors (SBCR) has received little attention, despite water being a major by-product. Although there have been numerous investigations to measure solubilities and mass transfer parameters of CO and H2 in F-T hydrocarbon fractions and slurries under a variety of operating conditions, the effect of water is typically neglected. Moreover, in most of the F-T SBCR models, liquid properties are typically determined for pure hydrocarbons or hydrocarbon mixtures, while disregarding the effect of water saturation on the physical properties of the hydrocarbons or on the gas solubilities. -

Capillary Condensation of Binary and Ternary Mixtures of N-Pentane
Article Cite This: Langmuir 2018, 34, 1967−1980 pubs.acs.org/Langmuir Capillary Condensation of Binary and Ternary Mixtures of n‑ − − Pentane Isopentane CO2 in Nanopores: An Experimental Study on the Effects of Composition and Equilibrium Elizabeth Barsotti,* Soheil Saraji, Sugata P. Tan, and Mohammad Piri Department of Petroleum Engineering, University of Wyoming, Laramie, Wyoming 82071, United States *S Supporting Information ABSTRACT: Confinement in nanopores can significantly impact the chemical and physical behavior of fluids. While some quantitative under- standing is available for how pure fluids behave in nanopores, there is little such insight for mixtures. This study aims to shed light on how nanoporosity impacts the phase behavior and composition of confined mixtures through comparison of the effects of static and dynamic equilibrium on experimentally measured isotherms and chromatographic analysis of the experimental fluids. To this end, a novel gravimetric apparatus is introduced and validated. Unlike apparatuses that have been previously used to study the confinement-induced phase behavior of fluids, this apparatus employs a gravimetric technique capable of discerning phase transitions in a wide variety of nanoporous media under both static and dynamic conditions. The apparatus was successfully validated against data in the literature for pure carbon dioxide and n-pentane. Then, isotherms were generated for binary mixtures of carbon dioxide and n- pentane using static and flow-through methods. Finally, two ternary mixtures of carbon dioxide, n-pentane, and isopentane were measured using the static method. While the equilibrium time was found important for determination of confined phase transitions, flow rate in the dynamic method was not found to affect the confined phase behavior. -

Flashbacks Causes and Prevention
Flashbacks Causes and Prevention Dan Banks, PE www.banksengineering.com Overview Flashbacks in process equipment are dangerous Best Practice: Design to avoid flashbacks Prediction: We have data and calculations to predict when a gas mixture is “flammable”. Avoid forming a flammable mixture to avoid problems. Worst Case: Equipment failure, operator error or “the unexpected” result in a flashback, anyway. Backup Plan: When there’s a chance that one day a flashback might occur, we build in countermeasures to keep them isolated and extinguish them quickly. This presentation explains how we predict flammability in various situations, and how special hardware is used to stop a flashback in its tracks. Not covered: hydrocarbon leaks to atmosphere, dust explosions, steam “explosions”, chemical reactions aside from combustion Flammability Flammable mixtures have enough oxygen and enough hydrocarbons to sustain combustion, once ignited. A mixture can be too “lean” or too “rich” to be flammable. The Lower Explosive Limit (LEL) of methane in air is 5% by volume. 4.9% methane in air will not sustain combustion at ambient temperature and pressure. The Upper Explosive Limit (UEL) of methane in air is 15%. Enriching to 15.1% makes the mixture non flammable at ambient temperature and pressure. Heating or pressurizing the methane/air mixture changes the LEL and UEL values (as described later). Next slide: tested values for a number of common hydrocarbons. Some Laboratory Values Hydrocarbon Formula LEL in air UEL in air Ignition (%) (%) Temperature, oF Methane CH4 5.0 15.0 1202 Ethane C2H6 3.0 12.4 959 Propane C3H8 2.1 9.5 871 n-Butane C4H10 1.8 8.4 896 n-Pentane C5H12 1.4 7.8 878 n-Hexane C6H14 1.2 7.4 527 n-Heptane C7H16 1.05 6.7 491 Dimethyl ether C2H6O 3.4 27 662 Hydrogen H2 4.0 75 1062 Ethylene oxide C2H4O 3.6 100 804 Acetylene C2H2 2.5 100 581 Ignition Temperature The table has a column for “ignition temperature” – a different value for each hydrocarbon. -

Final Updated Petroleum Hydrocarbon Fraction Toxicity Values for the Vph
FINAL UPDATED PETROLEUM HYDROCARBON FRACTION TOXICITY VALUES FOR THE VPH/EPH/APH METHODOLOGY Prepared for: Bureau of Waste Site Cleanup Massachusetts Department of Environmental Protection Boston, MA Prepared by: Office of Research and Standards Massachusetts Department of Environmental Protection Boston, MA November 2003 PREFACE In 1994 the Massachusetts Department of Environmental Protection (MA DEP) described a new approach for the evaluation of human health risks from ingestion exposures to complex petroleum hydrocarbon mixtures (MA DEP, 1994). The basis of the new approach was treating groups of compounds as if they had similar toxicities, in the absence of specific toxicity information on all the members of the group. The report included oral toxicity values for each of the designated petroleum hydrocarbon fractions. Since that time, there have been more recent efforts by others on this topic and additional information has become available to serve as a basis for updating the toxicity values. This document contains reviews of the more recent information, revisions to the oral toxicity values proposed in the 1994 report and new inhalation toxicity values. Readers will find different hydrocarbon compound size cutoffs for the hydrocarbon ranges identified here and in the 1994 report, compared to those in other related supporting documentation for this approach (i.e., the MA DEP VPH/EPH/APH analytical methods; guidance for Characterizing Risks Posed by Petroleum Contaminated Sites (MA DEP, 2002); risk spreadsheets for the MA DEP Bureau of Waste Site Cleanup). These differences relate primarily to subdivisions or truncation of ranges mandated by the analytical protocols employed for the analysis of the hydrocarbons (volatile (VPH), extractable (EPH) and air-phase(APH)) . -

US2688645.Pdf
Sept. 7, 1954 D. E. BADERTSCHER TAL 2,688,645 SOLVENT EXTRACTION Filed Sept. 10, 1952 4. Sheets-Sheet 4 AewZawe Aawza NE AvAyzewa (246OWA7a AA/AWe COWA/WWG 5% W47aa Aawza NE Af//YZEAE GAgaowana Aayawa cow/A/w/WG 622 GZYaoz. Aaw/W ANÉea AZAAAA WA4WC/6 BY GAOQ6A. C. JoAVsow & & ), e.g- Patented Sept. 7, 1954 2,688,645 UNITED STATES PATENT OFFICE 2,688,645 SOLVENT EXTRACTION Darwin E. Badertscher, Pitman, and Alfred W. Francis, and George C. Johnson, Woodbury, N. J., assignors to Socony-Wacuum Oil Com pany, incorporated, a corporation of New York Application September 10, 1952, Serial No. 308,828 19 Claims. (CI. 260-674) 1. 2 This invention is concerned With extraction and non-aromatic hydrocarbons, with a cyclic with certain selective solvents of . Various mix Organic carbonate. Such as ethylene carbonate, tures, and particularly of hydrocarbon mixtures, C2H4CO3, which is a cyclic ester represented by to separate the mixtures into fractions having the formula: different properties. EC-O Numerous processes have been developed for N Cs-O the separation of hydrocarbons and hydrocarbon / derivatives of different molecular configuration BC-O by taking advantage of their behaviour With As a class, the cyclic organic carbonates con selective agents. For example, aromaticS Such O templated herein are represented by the general as benzene, toluene and xylenes have been Sep formulae: arated from hydrocarbon mixtures in which they occur, by adsorption on gels such as Silica-alu 'R mina composites and the like, by azeotropic dis R-6-0 tillation, and by solvent extraction. -

Total Petroleum Hydrocarbons
TOTAL PETROLEUM HYDROCARONS 93 6. HEALTH EFFECTS 6.1 INTRODUCTION The primary purpose of this chapter is to provide public health officials, physicians, toxicologists, and other interested individuals and groups with an overall perspective on the toxicology of total petroleum hydrocarbons (TPH), and an understanding of various approaches used to assess petroleum hydrocarbons on the basis of fractions, individual indicator compounds, and appropriate surrogates. This chapter also provides descriptions and evaluations of toxicological studies and epidemiological investigations for these TPH fractions, indicator compounds, and surrogates, and provides conclusions, where possible, on the relevance of toxicity and toxicokinetics data to public health. 6.1.1 TPH Definition and Issues Overview. The assessment of petroleum hydrocarbon-contaminated sites has involved analysis for “total petroleum hydrocarbons” or TPH. TPH is a loosely defined aggregate that depends on the method of analysis as well as the contaminating material, and represents the total mass of hydrocarbons without identification of individual components (see Chapter 3). As TPH is not a consistent entity, the assessment of health effects and development of health guidance values, such as Minimal Risk Levels (MRLs) for TPH as a single entity are problematic. Earlier in the profile (Chapters 2 and 3), various TPH approaches were presented that divide TPH into fractions or groups of compounds based on analytical, fate and transport, and exposure issues. Similarly, several different approaches have also been evolving to assess the health effects of TPH on the basis of indicator compounds for separate fractions, which consist of petroleum hydrocarbons with similar physical and chemical properties. ATSDR’s approach to potential health effects from exposure to TPH uses surrogate health effects guidelines for each fraction, whether they represent an individual compound or a whole petroleum product. -
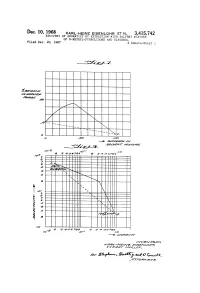
Nz Eisenlohr ET AL 3,415,742 RECOWERY of AROMATICS by EXTRACTION with SOLVENT MIXTURE of N-METHYL-PYRROLIDONE and GLYCEROL Filed Dec
Dec. 10, 1968 KARL-Heinz Eisenlohr ET AL 3,415,742 RECOWERY OF AROMATICS BY EXTRACTION WITH SOLVENT MIXTURE OF N-METHYL-PYRROLIDONE AND GLYCEROL Filed Dec. 20, 1967 3. Sheets-Sheet l 32-24 || | | | | | | | | | | | 1z-LMasa | | | | as a -N | | | | | -1 N | | | | | -- acaya aa 1/ - CZZM-2. acadalay1/7 Ma17Za Mo-a o1 a 6d 2 a 3 12 as as 299 a 3 a. pes. Ze 9 ?o a sizzaT N vo, a N 26 N 4 H N Med s H -- 2 2 17-ases 2ag.9 e as 12 attes Zage 24 o-W 16 //W1a/WYears aaZ 1/a/MZazaa/YZ2/4. A Ca2az M-742 / ae, as1 2Ape-, 742 al-4 o Gille. , 177 701/Way/cs Dec. 10, 1968 KARL-Heinz Eisenlohr ET AL 3,415,742 RECOVERY OF AROMATICS BY EXTRACTION WITH SOLVENT MIXTURE OF N-METHYL-PYRROLIDONE AND GLYCEROL Filed Dec. 20, 1967 3. Sheets-Sheet 2 AAA || aO // F Mao 17III I// /o ao so 170 af2 72e2. -> 2.6462% // - sa/A27W7M//Y70/ea MMW1a/Yatars 122-1/a/M/.4% sa/WZaa/at e-A-HA-1-1a cata/a/ auty- MYeaaaas, and 6 a.s.leaa 1772/71axs Dec. 10, 1968 KARL-Heinz Eisenlohr ETAL 3,415,742 RECOWERY OF AROMATICS BY EXTRACTION WITH SOLVENT MIXTURE OF N-METHYL-PYRROLIDONE AND GLYCEROL Filed Dec, 20, 1967 3. Sheets-Sheet 3 /V\ /VVN /VVVN /VVVX7M /VVAX7VNAN/A/YYYYA /Ya7 VVVVN Z27 VVVVVVVN 7VVVVVVVVN t es ad Yade aaraaya 17aeata/77. //W1aaware/re 12a-a-Me.at/sa/WZoast a ca1%7 /76 Zaates United States Patent Office Patented Dec.