Apoptosis in the Extraosseous Calcification Process
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sarcoidosis, Hypercalcemia and Calcium-Sensing Receptor Mutation: a Case Report
468 Letters to the Editor Sarcoidosis, Hypercalcemia and Calcium-sensing Receptor Mutation: A Case Report Shreya Dixit1 and Pablo Fernandez-Peñas1,2* 1Skin and Cancer Foundation Australia, 7 Ashley Lane, Westmead, NSW 2145, and 2Sydney Medical School Western, The University of Sydney, NSW, Australia. *E-mail: [email protected] Accepted October 28, 2010. Sarcoidosis is a multisystem granulomatous disease of The incidence of hypercalcaemia in patients with unknown aetiology. It predominantly affects the lungs; sarcoidosis is approximately 10% (3). It has previously however, 25% of patients have skin involvement, ranging been found that these abnormalities of calcium metabo- from non-specific maculopapular eruptions to erythema lism are due to dysregulated production of calcitriol by nodosum and lupus pernio (1). activated macrophages trapped in pulmonary alveoli and Familial hypocalciuric hypercalcaemia (FHH) results granulomatous inflammation (4). Conversion of calcidiol from an autosomal dominantly inherited inactivating to calcitriol is facilitated by 1 alpha-hydroxylase, a cyto- mutation of the calcium-sensing receptor (CaSR) gene chrome P450 enzyme. The activity of 1 alpha-hydroxyla- and is typically asymptomatic (2). Sarcoidosis has not se is tightly regulated through complex mechanisms that previously been reported in FHH. We had the opportu- depend on the circulating levels of calcium, phosphorus, nity to study a patient with sarcoidosis that has a CaSR PTH, 1,25 (OH)2D3 (calcitriol) and calcitonin. The role mutation. of CaSR in the regulation of 1 alpha-hydroxylase has not been defined, although there are suggestions that CaSR activation can repress the enzyme (5). CASE REPORT The incidence of FHH in patients with sarcoidosis A 70-year-old woman was referred by her endocrinologist with is unknown. -

Phytoplankton As Key Mediators of the Biological Carbon Pump: Their Responses to a Changing Climate
sustainability Review Phytoplankton as Key Mediators of the Biological Carbon Pump: Their Responses to a Changing Climate Samarpita Basu * ID and Katherine R. M. Mackey Earth System Science, University of California Irvine, Irvine, CA 92697, USA; [email protected] * Correspondence: [email protected] Received: 7 January 2018; Accepted: 12 March 2018; Published: 19 March 2018 Abstract: The world’s oceans are a major sink for atmospheric carbon dioxide (CO2). The biological carbon pump plays a vital role in the net transfer of CO2 from the atmosphere to the oceans and then to the sediments, subsequently maintaining atmospheric CO2 at significantly lower levels than would be the case if it did not exist. The efficiency of the biological pump is a function of phytoplankton physiology and community structure, which are in turn governed by the physical and chemical conditions of the ocean. However, only a few studies have focused on the importance of phytoplankton community structure to the biological pump. Because global change is expected to influence carbon and nutrient availability, temperature and light (via stratification), an improved understanding of how phytoplankton community size structure will respond in the future is required to gain insight into the biological pump and the ability of the ocean to act as a long-term sink for atmospheric CO2. This review article aims to explore the potential impacts of predicted changes in global temperature and the carbonate system on phytoplankton cell size, species and elemental composition, so as to shed light on the ability of the biological pump to sequester carbon in the future ocean. -
Reduced Calcification of Marine Plankton in Response to Increased
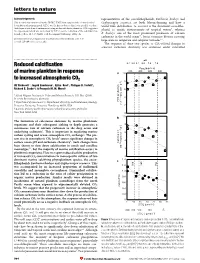
letters to nature Acknowledgements representatives of the coccolithophorids, Emiliania huxleyi and This research was sponsored by the EPSRC. T.W.F. ®rst suggested the electrochemical Gephyrocapsa oceanica, are both bloom-forming and have a deoxidation of titanium metal. G.Z.C. was the ®rst to observe that it was possible to reduce world-wide distribution. G. oceanica is the dominant coccolitho- thick layers of oxide on titanium metal using molten salt electrochemistry. D.J.F. suggested phorid in neritic environments of tropical waters9, whereas the experiment, which was carried out by G.Z.C., on the reduction of the solid titanium dioxide pellets. M. S. P. Shaffer took the original SEM image of Fig. 4a. E. huxleyi, one of the most prominent producers of calcium carbonate in the world ocean10, forms extensive blooms covering Correspondence and requests for materials should be addressed to D. J. F. large areas in temperate and subpolar latitudes9,11. (e-mail: [email protected]). The response of these two species to CO2-related changes in seawater carbonate chemistry was examined under controlled ................................................................. pH Reduced calci®cation 8.4 8.2 8.1 8.0 7.9 7.8 PCO2 (p.p.m.v.) of marine plankton in response 200 400 600 800 a 10 to increased atmospheric CO2 ) 8 –1 Ulf Riebesell *, Ingrid Zondervan*, BjoÈrn Rost*, Philippe D. Tortell², d –1 Richard E. Zeebe*³ & FrancËois M. M. Morel² 6 * Alfred Wegener Institute for Polar and Marine Research, P.O. Box 120161, 4 D-27515 Bremerhaven, Germany mol C cell –13 ² Department of Geosciences & Department of Ecology and Evolutionary Biology, POC production Princeton University, Princeton, New Jersey 08544, USA (10 2 ³ Lamont-Doherty Earth Observatory, Columbia University, Palisades, New York 10964, USA 0 ............................................................................................................................................. -

Distribution of Calcium Phosphate in the Exoskeleton of Larval Exeretonevra Angustifrons Hardy (Diptera: Xylophagidae)
Arthropod Structure & Development 34 (2005) 41–48 www.elsevier.com/locate/asd Distribution of calcium phosphate in the exoskeleton of larval Exeretonevra angustifrons Hardy (Diptera: Xylophagidae) Bronwen W. Cribba,b,*, Ron Rascha, John Barrya, Christopher M. Palmerb,1 aCentre for Microscopy and Microanalysis, The University of Queensland, Brisbane, Qd 4072, Australia bDepartment of Zoology and Entomology, The University of Queensland, Brisbane, Qd 4072, Australia Received 28 July 2004; accepted 26 August 2004 Abstract Distribution and organisation of the mineral, amorphous calcium phosphate (ACP), has been investigated in the exoskeleton of the xylophagid fly larva Exeretonevra angustifrons Hardy. While head capsule and anal plate are smooth with a thin epicuticle, the epicuticle of the body is thicker and shows unusual micro-architecture comprised of minute hemispherical (dome-shaped) protrusions. Electron microprobe analysis and energy dispersive spectroscopy revealed heterogeneity of mineral elements across body cuticle and a concentration of ACP in the epicuticle, especially associated with the hemispherical structures. Further imaging and analysis showed the bulk of the ACP to be present in nano-sized granules. It is hypothesised that the specific distribution of ACP may enhance cuticular hardness or durability without reducing flexibility. q 2004 Elsevier Ltd. All rights reserved. Keywords: Insect; Cuticle; Integument; Hardening; Analytical electron microscopy; Electron microprobe 1. Introduction was distributed heterogeneously. Further investigation of the distribution of the mineral phase at the micron and Strengthening of biological structures through cuticular nanometre level is needed to discover where deposition is calcification is well developed in decapod crustaceans but it occurring and how this might affect exoskeletal organis- rarely occurs in insects, where it is poorly understood ation. -

Effects of Ocean Acidification and Sea-Level Rise on Coral Reefs
Effects of Ocean Acidification and Sea-Level Rise on Coral Reefs Coral reefs are vital to the long-term to produce CaCO3, carbon dioxide (CO2), As CO2 increases in the atmosphere, viability of coastal societies, providing and water (H2O). Over time as these more is absorbed by the surface of the economic, recreational, and aesthetic organisms grow and die, their skeletons ocean, where it combines with seawater value from which coastal communities break down and become calcium carbon- to make a weak acid called carbonic thrive. Some of the services that coral ate sediments. These sediments fill in acid (H2CO3). This process, called reefs provide include protection from the framework of the reef and eventually ocean acidification, causes a decrease storm waves, nurseries and habitats for become cemented together, construct- in seawater pH (or increase in acidity) commercially important fish species, ing the foundation for continued upward that can result in a decrease in biogenic and production of sand for beaches. growth of the reef structure. The infilling calcification rates, dissolution of carbon- Coral reefs develop over thousands of the reef framework with sediments is ate sediments, and loss of reef structure. of years as tropical marine organ- what allows vertical accretion over time One of the primary concerns associated isms build skeletons of calcium carbonate and enables reef growth to keep up with with ocean acidification is whether coral (CaCO3) minerals to form a three-dimen- sea-level rise. Calcification is a revers- reefs will be able to continue to grow at sional structure (fig. 1). This process, ible process. -

Biomineralization and Evolutionary History Andrew H
1 111 Biomineralization and Evolutionary History Andrew H. Knoll Department of Organismic and Evolutionary Biology Harvard University Cambridge, Massachusetts, 02138 U.S.A. INTRODUCTION The Dutch ethologist Niko Tinbergen famously distinguished between proximal and ultimate explanations in biology. Proximally, biologists seek a mechanistic understanding of how organisms function; most of this volume addresses the molecular and physiological bases of biomineralization. But while much of biology might be viewed as a particularly interesting form of chemistry, it is more than that. Biology is chemistry with a history, requiring that proximal explanations be grounded in ultimate, or evolutionary, understanding. The physiological pathways by which organisms precipitate skeletal minerals and the forms and functions of the skeletons they fashion have been shaped by natural selection through geologic time, and all have constrained continuing evolution in skeleton-forming clades. In this chapter, I outline some major patterns of skeletal evolution inferred from phylogeny and fossils (Figure 1), highlighting ways that our improving mechanistic knowledge of biomineralization can help us to understand this evolutionary record (see Leadbetter and Riding 1986; Lowenstam and Weiner 1989; Carter 1990; and Simkiss and Wilbur 1989 for earlier reviews). Figure 1. A geologic time scale for the past 1000 million years, showing the principal time divisions used in Earth science and the timing of major evolutionary events discussed in this chapter. Earlier intervals of time—the Mesoproterozoic (1600–1000 million years ago) and Paleoproterozoic (2500– 1600 million years ago) eras of the Proterozoic Eon and the Archean Eon (> 2500 million years ago)— are not shown. Time scale after Remane (2000). -

A Novel Pharmacodynamic Assay to Evaluate the Effects of Crystallization
www.nature.com/scientificreports OPEN A novel pharmacodynamic assay to evaluate the efects of crystallization inhibitors on calcium Received: 1 March 2017 Accepted: 26 June 2017 phosphate crystallization in human Published online: 31 July 2017 plasma M. D. Ferrer1, M. M. Pérez1, M. M. Cànaves1, J. M. Buades 2, C. Salcedo1 & J. Perelló1,3 Cardiovascular calcifcation (CVC) is a progressive complication of chronic kidney disease and a predictor of CV events and mortality. The use of biomarkers to predict CV risk and activities of potential or current treatment drugs in these patients could have a crucial impact on therapeutic approaches. Our aim was to develop a novel assay for measurement of the rate of calcium phosphate crystallization in human plasma and provide a tool to evaluate the efects of crystallization inhibitors. The efcacy of inhibitors was determined by adding inhibitory compounds (polyphosphates, fetuin-A, sodium thiosulfate or citrate) to control samples. The assay was additionally validated for SNF472, an experimental formulation of phytate being developed for the treatment of calciphylaxis and CVC in patients with end-stage renal disease (ESRD) undergoing hemodialysis (HD). The method was repeatable and reproducible. The plasma crystallization rate was reduced up to 80% in a concentration-dependent manner following treatment with inhibitors in vitro, among which SNF472 was the most potent. This method appears benefcial in evaluating and discriminating between inhibitory activities of compounds such as polyphosphates on calcium phosphate crystallization, which present a novel therapeutic approach to treat CVC in ESRD patients. Calcifcation is the normal process of calcium salt deposition in body tissues occurring due to the presence of supersaturated or metastable salt solutions in biological fuids1, 2. -

The Cardiovascular System in Familial Hypocalciuric Hypercalcemia: A
175:4 N F B Jakobsen and others FHH and cardiovascular health 175:4 299–309 Clinical Study The cardiovascular system in familial hypocalciuric hypercalcemia: a cross- sectional study on physiological effects of inactivating variants in the calcium-sensing receptor gene Niels Frederik Breum Jakobsen1, Esben Laugesen1,2,3, Lars Rolighed4, Peter H Nissen5, Per Løgstrup Poulsen1, Erling Bjerregaard Pedersen3,6, Leif Mosekilde1 and Lars Rejnmark1,3 1Department of Endocrinology and Internal Medicine, Aarhus University Hospital, Aarhus, Denmark, Correspondence 2Danish Diabetes Academy, Odense University Hospital, Odense, Denmark, 3Department of Clinical should be addressed Medicine, Aarhus University, Aarhus, Denmark, 4Departments of Surgery and 5Clinical Biochemistry, to L Rejnmark Aarhus University Hospital, Aarhus, Denmark, and 6University Clinic in Nephrology and Hypertension, Email Holstebro Hospital, Hospital Jutland West, Holstebro, Denmark [email protected] Abstract Objective: Loss-of-function variants in the gene encoding the calcium-sensing receptor (CASR) result in familial hypocalciuric hypercalcemia (FHH), causing hypercalcemia with high normal or elevated parathyroid hormone levels. The CASR may also influence electrolyte and water homeostasis. It is unknown whether FHH affects cardiovascular health. We, therefore investigated whether FHH is associated with changes in the regulation of the cardiovascular system by measuring 24-h blood pressure (BP), arterial stiffness and vasoactive hormones. European Journal European of Endocrinology Design: Cross-sectional study comparing 50 patients with FHH to age- and gender-matched controls. Results: Studied subjects (69% women) had a mean age of 56 years. A similar number of patients and controls (33%) were on treatment with antihypertensive drugs. Overall, no differences were found between groups in 24-h ambulatory BP or pulse wave velocity. -

Role of Phosphate in Biomineralization
Henry Ford Health System Henry Ford Health System Scholarly Commons Endocrinology Articles Endocrinology and Metabolism 7-25-2020 Role of Phosphate in Biomineralization Sanjay Kumar Bhadada Sudhaker D. Rao Henry Ford Health System, [email protected] Follow this and additional works at: https://scholarlycommons.henryford.com/endocrinology_articles Recommended Citation Bhadada SK, and Rao SD. Role of Phosphate in Biomineralization. Calcif Tissue Int 2020. This Article is brought to you for free and open access by the Endocrinology and Metabolism at Henry Ford Health System Scholarly Commons. It has been accepted for inclusion in Endocrinology Articles by an authorized administrator of Henry Ford Health System Scholarly Commons. Calcifed Tissue International https://doi.org/10.1007/s00223-020-00729-9 REVIEW Role of Phosphate in Biomineralization Sanjay Kumar Bhadada1 · Sudhaker D. Rao2,3 Received: 31 March 2020 / Accepted: 14 July 2020 © Springer Science+Business Media, LLC, part of Springer Nature 2020 Abstract Inorganic phosphate is a vital constituent of cells and cell membranes, body fuids, and hard tissues. It is a major intracel- lular divalent anion, participates in many genetic, energy and intermediary metabolic pathways, and is important for bone health. Although we usually think of phosphate mostly in terms of its level in the serum, it is needed for many biological and structural functions of the body. Availability of adequate calcium and inorganic phosphate in the right proportions at the right place is essential for proper acquisition, biomineralization, and maintenance of mass and strength of the skeleton. The three specialized mineralized tissues, bones, teeth, and ossicles, difer from all other tissues in the human body because of their unique ability to mineralize, and the degree and process of mineralization in these tissues also difer to suit the specifc functions: locomotion, chewing, and hearing, respectively. -

11B As Monitor of Calcification Site Ph in Divergent Marine Calcifying Organisms Jill Sutton, Yi-Wei Liu, Justin B
δ11B as monitor of calcification site pH in divergent marine calcifying organisms Jill Sutton, Yi-Wei Liu, Justin B. Ries, Maxence Guillermic, Emmanuel Ponzevera, Robert A. Eagle To cite this version: Jill Sutton, Yi-Wei Liu, Justin B. Ries, Maxence Guillermic, Emmanuel Ponzevera, et al.. δ11B as monitor of calcification site pH in divergent marine calcifying organisms. Biogeosciences, European Geosciences Union, 2018, 15 (5), pp.1447-1467. 10.5194/bg-15-1447-2018. hal-02651007 HAL Id: hal-02651007 https://hal.archives-ouvertes.fr/hal-02651007 Submitted on 29 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Biogeosciences, 15, 1447–1467, 2018 https://doi.org/10.5194/bg-15-1447-2018 © Author(s) 2018. This work is distributed under the Creative Commons Attribution 3.0 License. δ11B as monitor of calcification site pH in divergent marine calcifying organisms Jill N. Sutton1, Yi-Wei Liu1,2, Justin B. Ries3, Maxence Guillermic2, Emmanuel Ponzevera4, and Robert A. Eagle1,5,6 1Université de Brest, UMR 6539 CNRS/UBO/IRD/Ifremer, LEMAR, IUEM, 29280, Plouzané, France 2Université de Brest, UMR 6539 CNRS/UBO/IRD/Ifremer, LGO, IUEM, 29280, Plouzané, France 3Department of Marine and Environmental Sciences, Marine Science Center, Northeastern University, 430 Nahant Rd, Nahant, MA 01908, USA 4Unité de Recherche Géosciences Marines, Ifremer, 29280, Plouzané, France 5Institute of the Environment and Sustainability, University of California, Los Angeles, LaKretz Hall, 619 Charles E Young Dr E no. -

Golgi-Associated Calcification in the Coccolithophorid Phytoflagellate Hymenomonas Carterae Daniel Charles Williams Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1972 Golgi-associated calcification in the coccolithophorid phytoflagellate Hymenomonas carterae Daniel Charles Williams Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Biology Commons Recommended Citation Williams, Daniel Charles, "Golgi-associated calcification in the coccolithophorid phytoflagellate Hymenomonas carterae " (1972). Retrospective Theses and Dissertations. 5874. https://lib.dr.iastate.edu/rtd/5874 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This dissertation was produced from a microfilm copy of the original document. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the original submitted. The following explanation of techniques is provided to help you understand markings or patterns which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting thru an image and duplicating adjacent pages to insure you complete continuity. 2. When an image on the film is obliterated with a large round black mark, it is an indication that the photographer suspected that the copy may have moved during exposure and thus cause a blurred image. -
Beachcombers Field Guide
Beachcombers Field Guide The Beachcombers Field Guide has been made possible through funding from Coastwest and the Western Australian Planning Commission, and the Department of Fisheries, Government of Western Australia. The project would not have been possible without our community partners – Friends of Marmion Marine Park and Padbury Senior High School. Special thanks to Sue Morrison, Jane Fromont, Andrew Hosie and Shirley Slack- Smith from the Western Australian Museum and John Huisman for editing the fi eld guide. FRIENDS OF Acknowledgements The Beachcombers Field Guide is an easy to use identifi cation tool that describes some of the more common items you may fi nd while beachcombing. For easy reference, items are split into four simple groups: • Chordates (mainly vertebrates – animals with a backbone); • Invertebrates (animals without a backbone); • Seagrasses and algae; and • Unusual fi nds! Chordates and invertebrates are then split into their relevant phylum and class. PhylaPerth include:Beachcomber Field Guide • Chordata (e.g. fi sh) • Porifera (sponges) • Bryozoa (e.g. lace corals) • Mollusca (e.g. snails) • Cnidaria (e.g. sea jellies) • Arthropoda (e.g. crabs) • Annelida (e.g. tube worms) • Echinodermata (e.g. sea stars) Beachcombing Basics • Wear sun protective clothing, including a hat and sunscreen. • Take a bottle of water – it can get hot out in the sun! • Take a hand lens or magnifying glass for closer inspection. • Be careful when picking items up – you never know what could be hiding inside, or what might sting you! • Help the environment and take any rubbish safely home with you – recycle or place it in the bin. Perth• Take Beachcomber your camera Fieldto help Guide you to capture memories of your fi nds.