Page 1 of 13 Pleasersc Do Not Advances Adjust Margins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metabolic Engineering of Escherichia Coli for Poly(3-Hydroxybutyrate)
Lin et al. Microb Cell Fact (2015) 14:185 DOI 10.1186/s12934-015-0369-3 RESEARCH Open Access Metabolic engineering of Escherichia coli for poly(3‑hydroxybutyrate) production via threonine bypass Zhenquan Lin1,2,3†, Yan Zhang1,2,3†, Qianqian Yuan1,2,3,4, Qiaojie Liu1,2,3, Yifan Li1,2,3, Zhiwen Wang1,2,3, Hongwu Ma4*, Tao Chen1,2,3,5* and Xueming Zhao1,2,3 Abstract Background: Poly(3-hydroxybutyrate) (PHB), have been considered to be good candidates for completely biode- gradable polymers due to their similar mechanical properties to petroleum-derived polymers and complete biodeg- radability. Escherichia coli has been used to simulate the distribution of metabolic fluxes in recombinant E. coli pro- ducing poly(3-hydroxybutyrate) (PHB). Genome-scale metabolic network analysis can reveal unexpected metabolic engineering strategies to improve the production of biochemicals and biofuels. Results: In this study, we reported the discovery of a new pathway called threonine bypass by flux balance analysis of the genome-scale metabolic model of E. coli. This pathway, mainly containing the reactions for threonine synthesis and degradation, can potentially increase the yield of PHB and other acetyl-CoA derived products by reutilizing the CO2 released at the pyruvate dehydrogenase step. To implement the threonine bypass for PHB production in E. coli, we deregulated the threonine and serine degradation pathway and enhanced the threonine synthesis, resulting in 2.23-fold improvement of PHB titer. Then, we overexpressed glyA to enhance the conversion of glycine to serine and activated transhydrogenase to generate NADPH required in the threonine bypass. -

Effect of Peptide Histidine Isoleucine on Water and Electrolyte Transport in the Human Jejunum
Gut: first published as 10.1136/gut.25.6.624 on 1 June 1984. Downloaded from Gut, 1984, 25, 624-628 Alimentary tract and pancreas Effect of peptide histidine isoleucine on water and electrolyte transport in the human jejunum K J MORIARTY, J E HEGARTY, K TATEMOTO, V MUTT, N D CHRISTOFIDES, S R BLOOM, AND J R WOOD From the Department of Gastroenterology, St Bartholomew's Hospital, London, The Liver Unit, King's College Hospital, London, Department ofMedicine, Hammersmith Hospital, London, and Department of Biochemistry, Karolinska Institute, Stockholm, Sweden SUMMARY Peptide histidine isoleucine, a 27 amino acid peptide with close amino acid sequence homology to vasoactive intestinal peptide and secretin, is distributed throughout the mammalian intestinal tract, where it has been localised to intramural neurones. An intestinal perfusion technique has been used to study the effect of intravenous peptide histidine isoleucine (44.5 pmol/kg/min) on water and electrolyte transport from a plasma like electrolyte solution in human jejunum in vivo. Peptide histidine isoleucine infusion produced peak plasma peptide histidine isoleucine concentrations in the range 2000-3000 pmolIl, flushing, tachycardia and a reduction in diastolic blood pressure. Peptide histidine isoleucine caused a significant inhibition of net absorption of water, sodium, potassium and bicarbonate and induced a net secretion of chloride, these changes being completely reversed during the post-peptide histidine isoleucine period. These findings suggest that endogenous peptide histidine isoleucine may participate in the neurohumoral regulation of water and electrolyte transport in the human jejunum. http://gut.bmj.com/ Peptide histidine isoleucine, isolated originally from INTESTINAL PERFUSION mammalian small intestine, is a 27-amino acid After an eight hour fast, each subject swallowed a peptide having close amino acid sequence homology double lumen intestinal perfusion tube, incorpo- to vasoactive intestinal peptide and secretin. -

Isoleucine, an Essential Amino Acid, Prevents Liver Metastases of Colon Cancer by Antiangiogenesis Kazumoto Murata1 and Masami Moriyama2
Research Article Isoleucine, an Essential Amino Acid, Prevents Liver Metastases of Colon Cancer by Antiangiogenesis Kazumoto Murata1 and Masami Moriyama2 1The First Department of Internal Medicine, Mie University School of Medicine, Tsu, Mie, Japan and 2Microbiology and Immunology, Keio University School of Medicine, Tokyo, Japan Abstract infection in the airways of cystic fibrosis (8), and susceptibility to salmonella infection in mouse intestinal tracts (9). In addition to In spite of recent advances in the treatment of colon cancer, h multiple liver metastases of colon cancer are still difficult to their direct antimicrobial activities, -defensins are strong treat. Some chemotherapeutic regimens have been reported to chemotactic factors for memory T cells and dendritic cells, be efficient, but there is a high risk of side effects associated suggesting that they also play an important role in acquired immunity (10–12). h-defensins are also inducible by inflammatory with these. Here, we show that isoleucine, an essential amino a h acid, prevents liver metastases in a mouse colon cancer cytokines, such as tumor necrosis factor- and interleukin-1 (13, 14). Recently, Fehlbaum et al. (15) reported that isoleucine metastatic model. Because isoleucine is a strong inducer of h B-defensin, we first hypothesized that it prevented liver and its analogues are highly specific inducers of -defensins. We thus originally hypothesized that isoleucine may contribute to metastases via the accumulation of dendritic cells or memory B tumor immunity through both innate and acquired immunity by T cells through up-regulation of -defensin. However, neither h B-defensin nor immunologic responses were induced by induction of -defensins. -
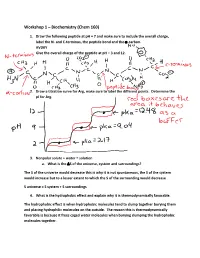
Workshop 1 – Biochemistry (Chem 160)
Workshop 1 – Biochemistry (Chem 160) 1. Draw the following peptide at pH = 7 and make sure to include the overall charge, label the N- and C-terminus, the peptide bond and the -carbon. AVDKY Give the overall charge of the peptide at pH = 3 and 12. 2. Draw a titration curve for Arg, make sure to label the different points. Determine the pI for Arg. 3. Nonpolar solute + water = solution a. What is the S of the universe, system and surroundings? The S of the universe would decrease this is why it is not spontaneous, the S of the system would increase but to a lesser extent to which the S of the surrounding would decrease S universe = S system + S surroundings 4. What is the hydrophobic effect and explain why it is thermodynamically favorable. The hydrophobic effect is when hydrophobic molecules tend to clump together burying them and placing hydrophilic molecules on the outside. The reason this is thermodynamically favorable is because it frees caged water molecules when burying clumping the hydrophobic molecules together. 5. Urea dissolves very readily in water, but the solution becomes very cold as the urea dissolves. How is this possible? Urea dissolves in water because when dissolving there is a net increase in entropy of the universe. The heat exchange, getting colder only reflects the enthalpy (H) component of the total energy change. The entropy change is high enough to offset the enthalpy component and to add up to an overall -G 6. A mutation that changes an alanine residue in the interior of a protein to valine is found to lead to a loss of activity. -

The Optimum Isoleucine:Lysine Ratio in Starter Diets to Maximize Growth Performance of the Early-Weaned Pig
Kansas Agricultural Experiment Station Research Reports Volume 0 Issue 10 Swine Day (1968-2014) Article 837 2000 The optimum isoleucine:lysine ratio in starter diets to maximize growth performance of the early-weaned pig B W. James Robert D. Goodband Michael D. Tokach See next page for additional authors Follow this and additional works at: https://newprairiepress.org/kaesrr Part of the Other Animal Sciences Commons Recommended Citation James, B W.; Goodband, Robert D.; Tokach, Michael D.; Nelssen, Jim L.; and DeRouchey, Joel M. (2000) "The optimum isoleucine:lysine ratio in starter diets to maximize growth performance of the early-weaned pig," Kansas Agricultural Experiment Station Research Reports: Vol. 0: Iss. 10. https://doi.org/10.4148/ 2378-5977.6677 This report is brought to you for free and open access by New Prairie Press. It has been accepted for inclusion in Kansas Agricultural Experiment Station Research Reports by an authorized administrator of New Prairie Press. Copyright 2000 Kansas State University Agricultural Experiment Station and Cooperative Extension Service. Contents of this publication may be freely reproduced for educational purposes. All other rights reserved. Brand names appearing in this publication are for product identification purposes only. No endorsement is intended, nor is criticism implied of similar products not mentioned. K-State Research and Extension is an equal opportunity provider and employer. The optimum isoleucine:lysine ratio in starter diets to maximize growth performance of the early-weaned pig Abstract A total of 360 weanling pigs (initially 12.3 lb BW and approximately 18 d of age) was used in a 14-d growth assay to determine the optimal isoleucine:lysine ratio to maximize growth performance. -

Cysteine Dioxygenase 1 Is a Metabolic Liability for Non-Small Cell Lung Cancer Authors: Yun Pyo Kang1, Laura Torrente1, Min Liu2, John M
bioRxiv preprint doi: https://doi.org/10.1101/459602; this version posted November 1, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer Authors: Yun Pyo Kang1, Laura Torrente1, Min Liu2, John M. Asara3,4, Christian C. Dibble5,6 and Gina M. DeNicola1,* Affiliations: 1 Department of Cancer Physiology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, USA 2 Proteomics and Metabolomics Core Facility, Moffitt Cancer Center and Research Institute, Tampa, FL, USA 3 Division of Signal Transduction, Beth Israel Deaconess Medical Center, Boston, MA, USA 4 Department of Medicine, Harvard Medical School, Boston, MA, USA 5 Department of Pathology and Cancer Center, Beth Israel Deaconess Medical Center, Boston, MA, USA 6 Department of Pathology, Harvard Medical School, Boston, MA, USA *Correspondence to: [email protected]. Keywords: KEAP1, NRF2, cysteine, CDO1, sulfite Summary NRF2 is emerging as a major regulator of cellular metabolism. However, most studies have been performed in cancer cells, where co-occurring mutations and tumor selective pressures complicate the influence of NRF2 on metabolism. Here we use genetically engineered, non-transformed primary cells to isolate the most immediate effects of NRF2 on cellular metabolism. We find that NRF2 promotes the accumulation of intracellular cysteine and engages the cysteine homeostatic control mechanism mediated by cysteine dioxygenase 1 (CDO1), which catalyzes the irreversible metabolism of cysteine to cysteine sulfinic acid (CSA). Notably, CDO1 is preferentially silenced by promoter methylation in non-small cell lung cancers (NSCLC) harboring mutations in KEAP1, the negative regulator of NRF2. -

1 Abietic Acid R Abrasive Silica for Polishing DR Acenaphthene M (LC
1 abietic acid R abrasive silica for polishing DR acenaphthene M (LC) acenaphthene quinone R acenaphthylene R acetal (see 1,1-diethoxyethane) acetaldehyde M (FC) acetaldehyde-d (CH3CDO) R acetaldehyde dimethyl acetal CH acetaldoxime R acetamide M (LC) acetamidinium chloride R acetamidoacrylic acid 2- NB acetamidobenzaldehyde p- R acetamidobenzenesulfonyl chloride 4- R acetamidodeoxythioglucopyranose triacetate 2- -2- -1- -β-D- 3,4,6- AB acetamidomethylthiazole 2- -4- PB acetanilide M (LC) acetazolamide R acetdimethylamide see dimethylacetamide, N,N- acethydrazide R acetic acid M (solv) acetic anhydride M (FC) acetmethylamide see methylacetamide, N- acetoacetamide R acetoacetanilide R acetoacetic acid, lithium salt R acetobromoglucose -α-D- NB acetohydroxamic acid R acetoin R acetol (hydroxyacetone) R acetonaphthalide (α)R acetone M (solv) acetone ,A.R. M (solv) acetone-d6 RM acetone cyanohydrin R acetonedicarboxylic acid ,dimethyl ester R acetonedicarboxylic acid -1,3- R acetone dimethyl acetal see dimethoxypropane 2,2- acetonitrile M (solv) acetonitrile-d3 RM acetonylacetone see hexanedione 2,5- acetonylbenzylhydroxycoumarin (3-(α- -4- R acetophenone M (LC) acetophenone oxime R acetophenone trimethylsilyl enol ether see phenyltrimethylsilyl... acetoxyacetone (oxopropyl acetate 2-) R acetoxybenzoic acid 4- DS acetoxynaphthoic acid 6- -2- R 2 acetylacetaldehyde dimethylacetal R acetylacetone (pentanedione -2,4-) M (C) acetylbenzonitrile p- R acetylbiphenyl 4- see phenylacetophenone, p- acetyl bromide M (FC) acetylbromothiophene 2- -5- -

Amino Acid Chemistry
Handout 4 Amino Acid and Protein Chemistry ANSC 619 PHYSIOLOGICAL CHEMISTRY OF LIVESTOCK SPECIES Amino Acid Chemistry I. Chemistry of amino acids A. General amino acid structure + HN3- 1. All amino acids are carboxylic acids, i.e., they have a –COOH group at the #1 carbon. 2. All amino acids contain an amino group at the #2 carbon (may amino acids have a second amino group). 3. All amino acids are zwitterions – they contain both positive and negative charges at physiological pH. II. Essential and nonessential amino acids A. Nonessential amino acids: can make the carbon skeleton 1. From glycolysis. 2. From the TCA cycle. B. Nonessential if it can be made from an essential amino acid. 1. Amino acid "sparing". 2. May still be essential under some conditions. C. Essential amino acids 1. Branched chain amino acids (isoleucine, leucine and valine) 2. Lysine 3. Methionine 4. Phenyalanine 5. Threonine 6. Tryptophan 1 Handout 4 Amino Acid and Protein Chemistry D. Essential during rapid growth or for optimal health 1. Arginine 2. Histidine E. Nonessential amino acids 1. Alanine (from pyruvate) 2. Aspartate, asparagine (from oxaloacetate) 3. Cysteine (from serine and methionine) 4. Glutamate, glutamine (from α-ketoglutarate) 5. Glycine (from serine) 6. Proline (from glutamate) 7. Serine (from 3-phosphoglycerate) 8. Tyrosine (from phenylalanine) E. Nonessential and not required for protein synthesis 1. Hydroxyproline (made postranslationally from proline) 2. Hydroxylysine (made postranslationally from lysine) III. Acidic, basic, polar, and hydrophobic amino acids A. Acidic amino acids: amino acids that can donate a hydrogen ion (proton) and thereby decrease pH in an aqueous solution 1. -

The Role of Some of the Krebs Cycle Reactions in the Biosynthetic Functions of Thiobacillus Thioparus
AN ABSTRACT OF THE THESIS OF Gerald G. Still for the PhD in Chemistry (Name) (Degree) (Major) Date thesis is presented May 14, 1965 Title THE ROLE OF SOME OF THE KREBS CYCLE REACTIONS IN THE BIOSYNTHETIC FUNCTIONS OF THIOBACILLUS THIOPARUS Abstract approved Redacted for Privacy (Major professor) Aseptic radiorespirometry has been used to examine the utilization of external carbon sources by proliferat- ing Thiobacillus thioparus cells. These studies reveal that glucose, galactose, mannose, fructose, ribose, DL- glutamate, and L- aspartate were not utilized by this chemoautotrophic organism. However, it has been shown that trace amounts of acetate, glycine, DL- serine, DL- alanine, succinate and fumarate can be utilized by T. thioparus cells. To elucidate the nature of the biosynthetic pathways operative in this bacteria, proliferating cell cultures were allowed to metabolize specifically 14C labeled substrates. The resulting 14C labeled cells were sub- sequently hydrolyzed, their amino acids isolated and subjected to degradation experiments. Examination of the respective fates of the label in DL- alanine- 2 -14C, acetate- 1 -14C, or acetate -2 -14C in the cellular metabolism revealed that the Krebs Cycle path- way is not functioning as a respiratory mechanism in T. thioparus. However, most of the reactions of the Krebs Cycle pathway are involved in the biosynthesis of carbon skeletons for various amino acids. A CO2 fixa- tion pathway of the C3 +C1 type is instrumental in provid- ing C4 dicarboxylic acids and those amino acids derived therefrom. Acetate can be incorporated into a -keto- glutarate and those amino acids derived therefrom, but cannot be incorporated into the C4 dicarboxylic acids. -

Biomoleculesbiomolecules
1414Unit Objectives BiomoleculesBiomolecules After studying this Unit, you will be able to • explain the characteristics of “It is the harmonious and synchronous progress of chemical biomolecules like carbohydrates, reactions in body which leads to life”. proteins and nucleic acids and hormones; • classify carbohydrates, proteins, A living system grows, sustains and reproduces itself. nucleic acids and vitamins on the The most amazing thing about a living system is that it basis of their structures; is composed of non-living atoms and molecules. The • explain the difference between pursuit of knowledge of what goes on chemically within DNA and RNA; a living system falls in the domain of biochemistry. Living • describe the role of biomolecules systems are made up of various complex biomolecules in biosystem. like carbohydrates, proteins, nucleic acids, lipids, etc. Proteins and carbohydrates are essential constituents of our food. These biomolecules interact with each other and constitute the molecular logic of life processes. In addition, some simple molecules like vitamins and mineral salts also play an important role in the functions of organisms. Structures and functions of some of these biomolecules are discussed in this Unit. 14.114.114.1 Carbohydrates Carbohydrates are primarily produced by plants and form a very large group of naturally occurring organic compounds. Some common examples of carbohydrates are cane sugar, glucose, starch, etc. Most of them have a general formula, Cx(H2O)y, and were considered as hydrates of carbon from where the name carbohydrate was derived. For example, the molecular formula of glucose (C6H12O6) fits into this general formula, C6(H2O)6. But all the compounds which fit into this formula may not be classified as carbohydrates. -

Mechanistic Study of Cysteine Dioxygenase, a Non-Heme
MECHANISTIC STUDY OF CYSTEINE DIOXYGENASE, A NON-HEME MONONUCLEAR IRON ENZYME by WEI LI Presented to the Faculty of the Graduate School of The University of Texas at Arlington in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY THE UNIVERSITY OF TEXAS AT ARLINGTON August 2014 Copyright © by Student Name Wei Li All Rights Reserved Acknowledgements I would like to thank Dr. Pierce for your mentoring, guidance and patience over the five years. I cannot go all the way through this without your help. Your intelligence and determination has been and will always be an example for me. I would like to thank my committee members Dr. Dias, Dr. Heo and Dr. Jonhson- Winters for the directions and invaluable advice. I also would like to thank all my lab mates, Josh, Bishnu ,Andra, Priyanka, Eleanor, you all helped me so I could finish my projects. I would like to thank the Department of Chemistry and Biochemistry for the help with my academic and career. At Last, I would like to thank my lovely wife and beautiful daughter who made my life meaningful and full of joy. July 11, 2014 iii Abstract MECHANISTIC STUDY OF CYSTEINE DIOXYGENASE A NON-HEME MONONUCLEAR IRON ENZYME Wei Li, PhD The University of Texas at Arlington, 2014 Supervising Professor: Brad Pierce Cysteine dioxygenase (CDO) is an non-heme mononuclear iron enzymes that catalyzes the O2-dependent oxidation of L-cysteine (Cys) to produce cysteine sulfinic acid (CSA). CDO controls cysteine levels in cells and is a potential drug target for some diseases such as Parkinson’s and Alzhermer’s. -

Importance of Acidic, Proline/Serine/Threonine-Rich, And
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 2501–2506, March 1997 Immunology Importance of acidic, prolineyserineythreonine-rich, and GTP- binding regions in the major histocompatibility complex class II transactivator: Generation of transdominant- negative mutants KEH-CHUANG CHIN*†,GEORGE G.-X. LI†‡, AND JENNY P.-Y. TING†‡ *Department of Biochemistry and Biophysics, †Lineberger Comprehensive Cancer Center, and ‡Department of Microbiology–Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7295 Communicated by George Stark, Cleveland Clinic Foundation, Cleveland, OH, December 31, 1996 (received for review September 3, 1996) ABSTRACT The class II transactivator (CIITA) is a (11). CIITA was cloned by its ability to complement RJ2.2.5, master transcription regulator of gene products involved in an in vitro-generated MHC class II negative cell derived from the exogenous antigen presentation pathway, including major Raji (11, 18). Several groups, including our own, have shown histocompatibility complex (MHC) class II, invariant chain, that CIITA is induced by IFN-g and that transfection of CIITA and DM. An extensive analysis of the putative functional alone into cells is sufficient to activate MHC class II (19–21), domains of CIITA is undertaken here to explore the action of invariant chain (19, 22), and HLA-DM genes (22). CIITA. Antibodies to CIITA protein were produced to verify A major issue in the field concerns the mode of action of that these mutant proteins are expressed. Both acidic and CIITA. Although CIITA is a strong transactivator, it does not prolineyserineythreonine-rich domains are essential for class bind MHC class II promoter elements, nor does it appear to II MHC promoter activation.