The Role of IFRD1 in the Recruitment and Function of Reserve Stem Cells in Regeneration and Cancer Mark Anthony Lewis Washington University in St
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Arp37342 T100
Aviva Systems Biology MEF2C antibody - N-terminal region (ARP37342_T100) Product Number ARP37342_T100 Product Page http://www.avivasysbio.com/mef2c-antibody-n-terminal-region-arp37342-t100.html Product Name MEF2C antibody - N-terminal region (ARP37342_T100) Size 100 ul Gene Symbol MEF2C Alias Symbols Mef2, AV011172, 5430401D19Rik, 9930028G15Rik Protein Size (# AA) 432 amino acids Molecular Weight 48kDa Product Format Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% sucrose. NCBI Gene Id 17260 Host Rabbit Clonality Polyclonal Concentration Batch dependent within range: 100 ul at 0.5 - 1 mg/ml Official Gene Full Myocyte enhancer factor 2C Name Description This is a rabbit polyclonal antibody against MEF2C. It was validated on Western Blot using a cell lysate as a positive control. Aviva Systems Biology strives to provide antibodies covering each member of a whole protein family of your interest. We also use our best efforts to provide you antibodies recognize various epitopes of a target protein. For availability of antibody needed for your experiment, please inquire ([email protected]). Peptide Sequence Synthetic peptide located within the following region: SRTNSDIVEALNKKENKGSESPDPDSSYALTPRTEEKYKKINEEFDNMIK Target Reference Shen,H., et al., (2006) Genes Dev. 20 (6), 675-688 Description of MEF2C is a transcription regulator of slow fiber Target Protein Interactions Vgll2; Hdac4; Nkx2-5; Hdac5; Phb2; KDM1A; Carm1; Ifrd1; Ncoa3; Ncoa2; Foxh1; Reconstitution and For short term use, store at 2-8C up to 1 week. For long term storage, store at -20C in Storage small aliquots to prevent freeze-thaw cycles. Lead Time Domestic: within 1-2 days delivery International: 1-2 days *** Required Wet/Dry Ice Surcharge will automatically be applied upon checkout for the shipment. -

Regulation of Neuronal Gene Expression and Survival by Basal NMDA Receptor Activity: a Role for Histone Deacetylase 4
The Journal of Neuroscience, November 12, 2014 • 34(46):15327–15339 • 15327 Cellular/Molecular Regulation of Neuronal Gene Expression and Survival by Basal NMDA Receptor Activity: A Role for Histone Deacetylase 4 Yelin Chen,1 Yuanyuan Wang,1 Zora Modrusan,3 Morgan Sheng,1 and Joshua S. Kaminker1,2 Departments of 1Neuroscience, 2Bioinformatics and Computational Biology, and 3Molecular Biology, Genentech Inc., South San Francisco, California 94080 Neuronal gene expression is modulated by activity via calcium-permeable receptors such as NMDA receptors (NMDARs). While gene expression changes downstream of evoked NMDAR activity have been well studied, much less is known about gene expression changes that occur under conditions of basal neuronal activity. In mouse dissociated hippocampal neuronal cultures, we found that a broad NMDAR antagonist, AP5, induced robust gene expression changes under basal activity, but subtype-specific antagonists did not. While some of the gene expression changes are also known to be downstream of stimulated NMDAR activity, others appear specific to basal NMDARactivity.ThegenesalteredbyAP5treatmentofbasalcultureswereenrichedforpathwaysrelatedtoclassIIahistonedeacetylases (HDACs), apoptosis, and synapse-related signaling. Specifically, AP5 altered the expression of all three class IIa HDACs that are highly expressed in the brain, HDAC4, HDAC5, and HDAC9, and also induced nuclear accumulation of HDAC4. HDAC4 knockdown abolished a subset of the gene expression changes induced by AP5, and led to neuronal death under -

Download Author Version (PDF)
Molecular BioSystems Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/molecularbiosystems Page 1 of 29 Molecular BioSystems Mutated Genes and Driver Pathways Involved in Myelodysplastic Syndromes—A Transcriptome Sequencing Based Approach Liang Liu1*, Hongyan Wang1*, Jianguo Wen2*, Chih-En Tseng2,3*, Youli Zu2, Chung-che Chang4§, Xiaobo Zhou1§ 1 Center for Bioinformatics and Systems Biology, Division of Radiologic Sciences, Wake Forest University Baptist Medical Center, Winston-Salem, NC 27157, USA. 2 Department of Pathology, the Methodist Hospital Research Institute, -

Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals
animals Review Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals Mohammadreza Mohammadabadi 1 , Farhad Bordbar 1,* , Just Jensen 2 , Min Du 3 and Wei Guo 4 1 Department of Animal Science, Faculty of Agriculture, Shahid Bahonar University of Kerman, Kerman 77951, Iran; [email protected] 2 Center for Quantitative Genetics and Genomics, Aarhus University, 8210 Aarhus, Denmark; [email protected] 3 Washington Center for Muscle Biology, Department of Animal Sciences, Washington State University, Pullman, WA 99163, USA; [email protected] 4 Muscle Biology and Animal Biologics, Animal and Dairy Science, University of Wisconsin-Madison, Madison, WI 53558, USA; [email protected] * Correspondence: [email protected] Simple Summary: Skeletal muscle mass is an important economic trait, and muscle development and growth is a crucial factor to supply enough meat for human consumption. Thus, understanding (candidate) genes regulating skeletal muscle development is crucial for understanding molecular genetic regulation of muscle growth and can be benefit the meat industry toward the goal of in- creasing meat yields. During the past years, significant progress has been made for understanding these mechanisms, and thus, we decided to write a comprehensive review covering regulators and (candidate) genes crucial for muscle development and growth in farm animals. Detection of these genes and factors increases our understanding of muscle growth and development and is a great help for breeders to satisfy demands for meat production on a global scale. Citation: Mohammadabadi, M.; Abstract: Farm-animal species play crucial roles in satisfying demands for meat on a global scale, Bordbar, F.; Jensen, J.; Du, M.; Guo, W. -

TIS7 and Skmc15 Regulate Adipocyte Differentiation and Intestinal Lipid Absorption
bioRxiv preprint doi: https://doi.org/10.1101/719922; this version posted July 30, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 TIS7 and SKMc15 Regulate Adipocyte Differentiation and Intestinal Lipid Absorption 1 1,13 1,14 1,15 2 2,16 2 Ilja Vietor*, Domagoj Cikes, Kati Piironen, Ronald Gstir, Ivan Tancevski, Philipp Eller, Egon Demetz, 3 4 5 6 7,8 9,10,11 Michael Hess, Volker Kuhn (†), Gerald Degenhart, Johannes Rainer, Jan Rozman, Martin Klingenspor, 7,8,12 1 1 Martin Hrabe de Angelis, Taras Valovka, Lukas A. Huber 1 2 3 4 Division of Cell Biology, Biocenter, Department of Internal Medicine II, Department of Histology, Department Trauma 5 6 Surgery, Department Radiology, Medical University Innsbruck, Austria, Institute for Biomedicine, Eurac Research, 7 Bolzano, Italy, German Mouse Clinic, Institute of Experimental Genetics, Helmholtz Zentrum München, German 8 Research Center for Environmental Health, Neuherberg, Germany, German Center for Diabetes Research (DZD), 9 Neuherberg, Germany, Chair of Molecular Nutritional Medicine, Technical University of Munich, School of Life 10 Sciences, Weihenstephan, Freising, Germany, EKFZ - Else Kröner Fresenius Center for Nutritional Medicine, 11 Technical University of Munich, Freising, Germany, ZIEL - Institute for Food & Health, Technical University of Munich, 12 Freising, Germany, Chair of Experimental Genetics, Technical University of Munich, School of Life Sciences, 13 Weihenstephan, Freising, Germany, IMBA, Institute of Molecular Biotechnology of the Austrian Academy of Sciences, 14 Vienna, Austria, Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, 15 16 Finland, ADSI – Austrian Drug Screening Institute GmbH, Innsbruck, Austria, Department of Internal Medicine, Medical University Graz, Austria * Address correspondence to: Ilja Vietor, Division of Cell Biology, Biocenter, Innsbruck Medical University, Innrain 80- 82, A-6020 Innsbruck, Austria. -

New Insights Into the Cellular Temporal Response to Proteostatic Stress
TOOLS AND RESOURCES New insights into the cellular temporal response to proteostatic stress Justin Rendleman1†, Zhe Cheng1†, Shuvadeep Maity1†, Nicolai Kastelic2, Mathias Munschauer2, Kristina Allgoewer1, Guoshou Teo1, Yun Bin Matteo Zhang1, Amy Lei1, Brian Parker1, Markus Landthaler2,3, Lindsay Freeberg4, Scott Kuersten4, Hyungwon Choi5,6, Christine Vogel1* 1Center for Genomics and Systems Biology, Department of Biology, New York University, New York, United States; 2Berlin Institute for Medical Systems Biology, Max Delbru¨ ck Center for Molecular Medicine, Berlin, Germany; 3Integrative Research Institute for the Life Sciences, Institute of Biology, Humboldt University, Berlin, Germany; 4Illumina Inc, Madison, United States; 5National University of Singapore, Singapore; 6Institute of Molecular and Cell Biology, Agency for Science, Technology and Research, Singapore Abstract Maintaining a healthy proteome involves all layers of gene expression regulation. By quantifying temporal changes of the transcriptome, translatome, proteome, and RNA-protein interactome in cervical cancer cells, we systematically characterize the molecular landscape in response to proteostatic challenges. We identify shared and specific responses to misfolded proteins and to oxidative stress, two conditions that are tightly linked. We reveal new aspects of the unfolded protein response, including many genes that escape global translation shutdown. A subset of these genes supports rerouting of energy production in the mitochondria. We also find that many genes change at multiple levels, in either the same or opposing directions, and at different time points. We highlight a variety of putative regulatory pathways, including the stress- dependent alternative splicing of aminoacyl-tRNA synthetases, and protein-RNA binding within the 3’ untranslated region of molecular chaperones. These results illustrate the potential of this information-rich resource. -

The IFRD1 (57460C>T Polymorphism) Gene
a ular nd G ec en l e o t i M c f M o l e Journal of Molecular and Genetic d a i de Lima Marson et al., J Mol Genet Med 2013, 7:2 n c r i n u e o DOI: 10.4172/1747-0862.1000058 J Medicine ISSN: 1747-0862 Research Article Article OpenOpen Access Access The IFRD1 (57460C>T Polymorphism) Gene: A Negative Report in Cystic Fibrosis Clinical Severity Fernando Augusto de Lima Marson1,2*, Aline Roberta Bariani Marcelino1, Luciana Montes Rezende1, Antônio Fernando Ribeiro2, José Dirceu Ribeiro2 and Carmen Sílvia Bertuzzo1 1Department of Medical Genetics, University of Campinas, Unicamp, School of Medical Sciences, FCM, Tessália Vieira de Camargo, 126, Cidade Universitária "Zeferino Vaz", Campinas, SP, Brazil 2Department of Pediatrics, University of Campinas, Unicamp, School of Medical Sciences, FCM, Tessália Vieira de Camargo, 126, Cidade Universitária "Zeferino Vaz", Campinas, SP, Brazil Abstract Cystic fibrosis (CF) is an autosomal recessive disease caused by more than 1,900 mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. In CF, one intriguing aspect is that patients, with same CFTR mutation, can have high clinical variability. Thus, the CFTR genotype does not seem to be the only determining factor in the clinical severity modulation. Therefore, the modifier genes and the environment must be considered. The IFRD1 (Interferon-related developmental regulator 1) gene, acts on the immune system and in the recruitment of immune cells, and consequently could be a modulator. In our data we included 88 CF patients, diagnosed by CFTR mutation screening and positive sweat test. -

Oxidized Phospholipids Regulate Amino Acid Metabolism Through MTHFD2 to Facilitate Nucleotide Release in Endothelial Cells
ARTICLE DOI: 10.1038/s41467-018-04602-0 OPEN Oxidized phospholipids regulate amino acid metabolism through MTHFD2 to facilitate nucleotide release in endothelial cells Juliane Hitzel1,2, Eunjee Lee3,4, Yi Zhang 3,5,Sofia Iris Bibli2,6, Xiaogang Li7, Sven Zukunft 2,6, Beatrice Pflüger1,2, Jiong Hu2,6, Christoph Schürmann1,2, Andrea Estefania Vasconez1,2, James A. Oo1,2, Adelheid Kratzer8,9, Sandeep Kumar 10, Flávia Rezende1,2, Ivana Josipovic1,2, Dominique Thomas11, Hector Giral8,9, Yannick Schreiber12, Gerd Geisslinger11,12, Christian Fork1,2, Xia Yang13, Fragiska Sigala14, Casey E. Romanoski15, Jens Kroll7, Hanjoong Jo 10, Ulf Landmesser8,9,16, Aldons J. Lusis17, 1234567890():,; Dmitry Namgaladze18, Ingrid Fleming2,6, Matthias S. Leisegang1,2, Jun Zhu 3,4 & Ralf P. Brandes1,2 Oxidized phospholipids (oxPAPC) induce endothelial dysfunction and atherosclerosis. Here we show that oxPAPC induce a gene network regulating serine-glycine metabolism with the mitochondrial methylenetetrahydrofolate dehydrogenase/cyclohydrolase (MTHFD2) as a cau- sal regulator using integrative network modeling and Bayesian network analysis in human aortic endothelial cells. The cluster is activated in human plaque material and by atherogenic lipo- proteins isolated from plasma of patients with coronary artery disease (CAD). Single nucleotide polymorphisms (SNPs) within the MTHFD2-controlled cluster associate with CAD. The MTHFD2-controlled cluster redirects metabolism to glycine synthesis to replenish purine nucleotides. Since endothelial cells secrete purines in response to oxPAPC, the MTHFD2- controlled response maintains endothelial ATP. Accordingly, MTHFD2-dependent glycine synthesis is a prerequisite for angiogenesis. Thus, we propose that endothelial cells undergo MTHFD2-mediated reprogramming toward serine-glycine and mitochondrial one-carbon metabolism to compensate for the loss of ATP in response to oxPAPC during atherosclerosis. -

The Interferon-Related Developmental Regulator 1 Is Used by Human Papillomavirus to Suppress Nfkb Activation
ARTICLE Received 8 Sep 2014 | Accepted 5 Feb 2015 | Published 13 Mar 2015 DOI: 10.1038/ncomms7537 OPEN The interferon-related developmental regulator 1 is used by human papillomavirus to suppress NFkB activation Bart Tummers1, Renske Goedemans1, Laetitia P.L. Pelascini2, Ekaterina S. Jordanova3, Edith M.G. van Esch4, Craig Meyers5, Cornelis J.M. Melief6, Judith M. Boer7,w & Sjoerd H. van der Burg1 High-risk human papillomaviruses (hrHPVs) infect keratinocytes and successfully evade host immunity despite the fact that keratinocytes are well equipped to respond to innate and adaptive immune signals. Using non-infected and freshly established or persistent hrHPV- infected keratinocytes we show that hrHPV impairs the acetylation of NFkB/RelA K310 in keratinocytes. As a consequence, keratinocytes display a decreased pro-inflammatory cytokine production and immune cell attraction in response to stimuli of the innate or adaptive immune pathways. HPV accomplishes this by augmenting the expression of interferon-related developmental regulator 1 (IFRD1) in an EGFR-dependent manner. Restoration of NFkB/RelA acetylation by IFRD1 shRNA, cetuximab treatment or the HDAC1/3 inhibitor entinostat increases basal and induced cytokine expression. Similar observations are made in IFRD1-overexpressing HPV-induced cancer cells. Thus, our study reveals an EGFR–IFRD1-mediated viral immune evasion mechanism, which can also be exploited by cancer cells. 1 Department of Clinical Oncology, Leiden University Medical Center, Albinusdreef 2, 2333ZA Leiden, The Netherlands. 2 Department of Molecular Cell Biology, Leiden University Medical Center, Albinusdreef 2, 2333ZA Leiden, The Netherlands. 3 Center for Gynaecological Oncology, Plesmanlaan 121, 1066CX Amsterdam, The Netherlands. 4 Department of Gynaecology, Leiden University Medical Center, Albinusdreef 2, 2333ZA Leiden, The Netherlands. -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV). -
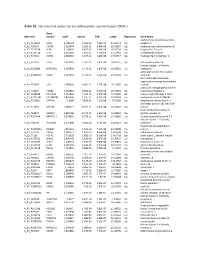
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Transcription Factor TEAD4 Regulates Expression of Myogenin and the Unfolded Protein Response Genes During C2C12 Cell Differentiation
Cell Death and Differentiation (2012) 19, 220–231 & 2012 Macmillan Publishers Limited All rights reserved 1350-9047/12 www.nature.com/cdd Transcription factor TEAD4 regulates expression of Myogenin and the unfolded protein response genes during C2C12 cell differentiation A Benhaddou1, C Keime1,TYe1, A Morlon1, I Michel1, B Jost1, G Mengus1 and I Davidson*,1 The TEAD (1–4) transcription factors comprise the conserved TEA/ATTS DNA-binding domain recognising the MCAT element in the promoters of muscle-specific genes. Despite extensive genetic analysis, the function of TEAD factors in muscle differentiation has proved elusive due to redundancy among the family members. Expression of the TEA/ATTS DNA-binding domain that acts as a dominant negative repressor of TEAD factors in C2C12 myoblasts inhibits their differentiation, whereas selective shRNA knockdown of TEAD4 results in abnormal differentiation characterised by the formation of shortened myotubes. Chromatin immunoprecipitation coupled to array hybridisation shows that TEAD4 occupies 867 promoters including those of myogenic miRNAs. We show that TEAD factors directly induce Myogenin, CDKN1A and Caveolin 3 expression to promote myoblast differentiation. RNA-seq identifies a set of genes whose expression is strongly reduced upon TEAD4 knockdown among which are structural and regulatory proteins and those required for the unfolded protein response. In contrast, TEAD4 represses expression of the growth factor CTGF (connective tissue growth factor) to promote differentiation. Together these