Identification of Common Predisposing Loci in Several Hematopoietic Cancers and Several Dog Breeds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SPNS2 Antibody (N-Term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap17856a
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 SPNS2 Antibody (N-term) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP17856a Specification SPNS2 Antibody (N-term) - Product Information Application WB, IHC-P, FC,E Primary Accession Q8IVW8 Other Accession NP_001118230.1 Reactivity Human Host Rabbit Clonality Polyclonal Isotype Rabbit IgG Antigen Region 68-94 SPNS2 Antibody (N-term) - Additional Information Gene ID 124976 Other Names Protein spinster homolog 2, SPNS2 All lanes : Anti-SPNS2 Antibody (N-term) at Target/Specificity 1:1000 dilution Lane 1: human brain tissue This SPNS2 antibody is generated from lysate Lane 2: 293 whole cell lysate Lane 3: rabbits immunized with a KLH conjugated SK-BR-3 whole cell lysate Lysates/proteins at synthetic peptide between 68-94 amino 20 µg per lane. Secondary Goat Anti-Rabbit acids from the N-terminal region of human IgG, (H+L), Peroxidase conjugated at 1/10000 SPNS2. dilution. Predicted band size : 58 kDa Blocking/Dilution buffer: 5% NFDM/TBST. Dilution WB~~1:1000 IHC-P~~1:100 FC~~1:25 Format Purified polyclonal antibody supplied in PBS with 0.09% (W/V) sodium azide. This antibody is purified through a protein A column, followed by peptide affinity purification. Storage Maintain refrigerated at 2-8°C for up to 2 weeks. For long term storage store at -20°C in small aliquots to prevent freeze-thaw cycles. Precautions SPNS2 Antibody (N-term) is for research use All lanes : Anti-SPNS2 Antibody (N-term) at only and not for use in diagnostic or 1:1000 dilution Lane 1: Human heart lysate therapeutic procedures. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

An Interaction Map of Circulating Metabolites, Immune Gene Networks, and Their Genetic Regulation Artika P
Nath et al. Genome Biology (2017) 18:146 DOI 10.1186/s13059-017-1279-y RESEARCH Open Access An interaction map of circulating metabolites, immune gene networks, and their genetic regulation Artika P. Nath1,2, Scott C. Ritchie2,3, Sean G. Byars3,4, Liam G. Fearnley3,4, Aki S. Havulinna5,6, Anni Joensuu5, Antti J. Kangas7, Pasi Soininen7,8, Annika Wennerström5, Lili Milani9, Andres Metspalu9, Satu Männistö5, Peter Würtz7,10, Johannes Kettunen5,7,8,11, Emma Raitoharju12, Mika Kähönen13, Markus Juonala14,15, Aarno Palotie6,16,17,18, Mika Ala-Korpela7,8,11,19,20, Samuli Ripatti6,21, Terho Lehtimäki12, Gad Abraham2,3,4, Olli Raitakari22,23, Veikko Salomaa5, Markus Perola5,6,9 and Michael Inouye1,2,3,4* Abstract Background: Immunometabolism plays a central role in many cardiometabolic diseases. However, a robust map of immune-related gene networks in circulating human cells, their interactions with metabolites, and their genetic control is still lacking. Here, we integrate blood transcriptomic, metabolomic, and genomic profiles from two population-based cohorts (total N = 2168), including a subset of individuals with matched multi-omic data at 7-year follow-up. Results: We identify topologically replicable gene networks enrichedfordiverseimmunefunctions including cytotoxicity, viral response, B cell, platelet, neutrophil, and mast cell/basophil activity. These immune gene modules show complex patterns of association with 158 circulating metabolites, including lipoprotein subclasses, lipids, fatty acids, amino acids, small molecules, and CRP. Genome-wide scans for module expression quantitative trait loci (mQTLs) reveal five modules with mQTLs that have both cis and trans effects. The strongest mQTL is in ARHGEF3 (rs1354034) and affects a module enriched for platelet function, independent of platelet counts. -

Mapping the Genetic Architecture of Gene Regulation in Whole Blood
Mapping the Genetic Architecture of Gene Regulation in Whole Blood Katharina Schramm1,2., Carola Marzi3,4,5., Claudia Schurmann6., Maren Carstensen7,8., Eva Reinmaa9,10, Reiner Biffar11, Gertrud Eckstein1, Christian Gieger12, Hans-Jo¨ rgen Grabe13, Georg Homuth6, Gabriele Kastenmu¨ ller14, Reedik Ma¨gi10, Andres Metspalu9,10, Evelin Mihailov10,15, Annette Peters2, Astrid Petersmann16, Michael Roden7,8,17, Konstantin Strauch12,18, Karsten Suhre14,19, Alexander Teumer6,UweVo¨ lker6, Henry Vo¨ lzke20, Rui Wang-Sattler3,4, Melanie Waldenberger3,4, Thomas Meitinger1,2,21, Thomas Illig22, Christian Herder7,8., Harald Grallert3,4,5., Holger Prokisch1,2*. 1 Institute of Human Genetics, Helmholtz Center Munich, German Research Center for Environmental Health, Neuherberg, Germany, 2 Institute of Human Genetics, Technical University Munich, Mu¨nchen, Germany, 3 Research Unit of Molecular Epidemiology, Helmholtz Center Munich, German Research Center for Environmental Health, Neuherberg, Germany, 4 Institute of Epidemiology II, Helmholtz Center Munich, German Research Center for Environmental Health, Neuherberg, Germany, 5 German Center for Diabetes Research (DZD e.V.), Neuherberg, Germany, 6 Interfaculty Institute for Genetics and Functional Genomics, Department of Functional Genomics, University Medicine Greifswald, Greifswald, Germany, 7 Institute for Clinical Diabetology, German Diabetes Center, Leibniz Center for Diabetes Research at Heinrich Heine University Du¨sseldorf, Du¨sseldorf, Germany, 8 German Center for Diabetes Research (DZD e.V.), -

Redefining the Specificity of Phosphoinositide-Binding by Human
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.20.163253; this version posted June 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Redefining the specificity of phosphoinositide-binding by human PH domain-containing proteins Nilmani Singh1†, Adriana Reyes-Ordoñez1†, Michael A. Compagnone1, Jesus F. Moreno Castillo1, Benjamin J. Leslie2, Taekjip Ha2,3,4,5, Jie Chen1* 1Department of Cell & Developmental Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801; 2Department of Biophysics and Biophysical Chemistry, Johns Hopkins University School of Medicine, Baltimore, MD 21205; 3Department of Biophysics, Johns Hopkins University, Baltimore, MD 21218; 4Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD 21205; 5Howard Hughes Medical Institute, Baltimore, MD 21205, USA †These authors contributed equally to this work. *Correspondence: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2020.06.20.163253; this version posted June 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. ABSTRACT Pleckstrin homology (PH) domains are presumed to bind phosphoinositides (PIPs), but specific interaction with and regulation by PIPs for most PH domain-containing proteins are unclear. Here we employed a single-molecule pulldown assay to study interactions of lipid vesicles with full-length proteins in mammalian whole cell lysates. -

Targeting the Methyltransferase SETD8 Impairs Tumor Cell Survival and Overcomes Drug Resistance Independently of P53 Status in Multiple Myeloma
bioRxiv preprint doi: https://doi.org/10.1101/776930; this version posted September 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Targeting the methyltransferase SETD8 impairs tumor cell survival and overcomes drug resistance independently of p53 status in multiple myeloma Laurie Herviou (1,2,4), Fanny Izard (3,4), Ouissem Karmous-Gadacha (2) , Claire Gourzones (1), Celine Bellanger (1), Eva Desmedt (5), Anqi Ma (6), Laure Vincent (7) , Guillaume Cartron (4,7), Karin Vanderkerken (5), Jian Jin (6), Elke De Bruyne (5), Charlotte Grimaud (3,4,8), Eric Julien (3,4,8 +) and Jérôme Moreaux (1,2,4+) (1) IGH, CNRS, Univ Montpellier, France (2) CHU Montpellier, Laboratory for Monitoring Innovative Therapies, Department of Biological Hematology, Montpellier, France (3) Institut de Recherche en Cancérologie de Montpellier (IRCM), INSERM U1194, Institut Régional du Cancer (ICM), Montpellier F-34298, France (4) University of Montpellier, Montpellier, F-34090, France (5) Department of Hematology and Immunology-Myeloma Center Brussels, Vrije Universiteit Brussel, Brussels, Belgium (6) Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, NeW York, NeW York 10029, United States. (7) CHU Montpellier, Department of Clinical Hematology, Montpellier, France (8) Centre National de la Recherche Scientifique (CNRS), F-34293, Montpellier, France + : co-last and corresponding authors ; corresponding authors: Eric Julien ([email protected]) and jérôme Moreaux ([email protected]). 1 bioRxiv preprint doi: https://doi.org/10.1101/776930; this version posted September 20, 2019. -

TRCP Promotes Cell Growth by Targeting PR-Set7/Set8 for Degradation
SCFβ-TRCP promotes cell growth by targeting PR-Set7/Set8 for degradation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Wang, Z., X. Dai, J. Zhong, H. Inuzuka, L. Wan, X. Li, L. Wang, et al. 2015. “SCFβ-TRCP promotes cell growth by targeting PR-Set7/Set8 for degradation.” Nature Communications 6 (1): 10185. doi:10.1038/ ncomms10185. http://dx.doi.org/10.1038/ncomms10185. Published Version doi:10.1038/ncomms10185 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:23993465 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ARTICLE Received 4 May 2015 | Accepted 12 Nov 2015 | Published 15 Dec 2015 DOI: 10.1038/ncomms10185 OPEN SCFb-TRCP promotes cell growth by targeting PR-Set7/Set8 for degradation Zhiwei Wang1,2,*, Xiangpeng Dai2,*, Jiateng Zhong2,3,*, Hiroyuki Inuzuka2, Lixin Wan2, Xiaoning Li2,4, Lixia Wang1, Xiantao Ye1, Liankun Sun4, Daming Gao2,5,LeeZou6 & Wenyi Wei2 The Set8/PR-Set7/KMT5a methyltransferase plays critical roles in governing transcriptional regulation, cell cycle progression and tumorigenesis. Although CRL4Cdt2 was reported to regulate Set8 stability, deleting the PIP motif only led to partial resistance to ultraviolet- induced degradation of Set8, indicating the existence of additional E3 ligase(s) controlling Set8 stability. Furthermore, it remains largely undefined how DNA damage-induced kinase cascades trigger the timely destruction of Set8 to govern tumorigenesis. -

Function in Immune System Spns2 Transporter the Role of Sphingosine-1-Phosphate
The Journal of Immunology The Role of Sphingosine-1-Phosphate Transporter Spns2 in Immune System Function Anastasia Nijnik,*,† Simon Clare,* Christine Hale,* Jing Chen,* Claire Raisen,* Lynda Mottram,* Mark Lucas,* Jeanne Estabel,* Edward Ryder,* Hibret Adissu,‡ Sanger Mouse Genetics Project,1 Niels C. Adams,* Ramiro Ramirez-Solis,* Jacqueline K. White,* Karen P. Steel,* Gordon Dougan,* and Robert E. W. Hancock† Sphingosine-1-phosphate (S1P) is lipid messenger involved in the regulation of embryonic development, immune system functions, and many other physiological processes. However, the mechanisms of S1P transport across cellular membranes remain poorly understood, with several ATP-binding cassette family members and the spinster 2 (Spns2) member of the major facilitator superfamily known to mediate S1P transport in cell culture. Spns2 was also shown to control S1P activities in zebrafish in vivo and to play a critical role in zebrafish cardiovascular development. However, the in vivo roles of Spns2 in mammals and its involvement in the different S1P-dependent physiological processes have not been investigated. In this study, we charac- terized Spns2-null mouse line carrying the Spns2tm1a(KOMP)Wtsi allele (Spns2tm1a). The Spns2tm1a/tm1a animals were viable, indi- cating a divergence in Spns2 function from its zebrafish ortholog. However, the immunological phenotype of the Spns2tm1a/tm1a mice closely mimicked the phenotypes of partial S1P deficiency and impaired S1P-dependent lymphocyte trafficking, with a depletion of lymphocytes in circulation, an increase in mature single-positive T cells in the thymus, and a selective reduction in mature B cells in the spleen and bone marrow. Spns2 activity in the nonhematopoietic cells was critical for normal lymphocyte development and localization. -
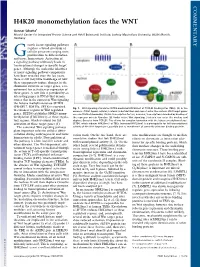
H4K20 Monomethylation Faces the WNT
COMMENTARY H4K20 monomethylation faces the WNT Gunnar Schotta1 Munich Center for Integrated Protein Science and Adolf Butenandt Institute, Ludwig Maximilians University, 80336 Munich, Germany rowth factor signaling pathways regulate a broad spectrum of G cellular processes ranging from proliferation to differentiation and tissue homeostasis. Activation of a signaling pathway ultimately leads to transcriptional changes in specific target genes. Although the molecular identities of many signaling pathway components have been revealed over the last years, there is still very little knowledge of how these components induce changes in the chromatin structure of target genes, a re- quirement for activation or repression of these genes. A new link is provided by an interesting paper in PNAS that demon- strates that in the context of Wnt signaling the histone methyltransferase SETD8 (PR-SET7, KMT5a, SET8) is recruited Fig. 1. Wnt signaling stimulates SETD8-mediated H4K20me1 at TCF/LEF binding sites (TBEs). (A)Inthe to enhancer regions of Wnt-regulated absence of Wnt ligand, cellular β-catenin is destabilized and cannot enter the nucleus. Wnt target genes genes. SETD8 establishes H4K20 mono- are constitutively bound by TCF/LEF transcription factors; however, transcription is blocked by binding of methylation (H4K20me1) at these regula- the repressor protein Groucho. (B) Under active Wnt signaling, β-catenin can enter the nucleus and tory regions, which is crucial for full displace Groucho from TCF/LEF. This allows for complex formation with the histone methyltransferase activation of these target genes (1). SETD8, which induces H4K20me1 at TBEs. Increased H4K20me1 is a prerequisite for full transcriptional The canonical Wnt signaling pathway activity of the Wnt target gene, possibly due to recruitment of currently unknown binding proteins. -

Mechanism of CRL4 , a PCNA-Dependent E3 Ubiquitin Ligase
Downloaded from genesdev.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Mechanism of CRL4Cdt2, a PCNA-dependent E3 ubiquitin ligase Courtney G. Havens and Johannes C. Walter1 Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, USA Eukaryotic cell cycle transitions are driven by E3 ubiq- the ubiquitin is transferred from E1 to the cysteine of an uitin ligases that catalyze the ubiquitylation and destruc- ‘‘E2’’ ubiquitin-conjugating enzyme. Finally, the E2 in- tion of specific protein targets. For example, the anaphase- teracts with an ‘‘E3’’ ubiquitin ligase that also binds the promoting complex/cyclosome (APC/C) promotes the substrate. The juxtaposition of the substrate and the charged exit from mitosis via destruction of securin and mitotic E2 enzyme leads to ubiquitin transfer to the substrate. cyclins, whereas CRL1Skp2 allows entry into S phase by The specificity of ubiquitylation is encoded at the level of targeting the destruction of the cyclin-dependent kinase substrate recognition by the E3 enzymes (Ravid and (CDK) inhibitor p27. Recently, an E3 ubiquitin ligase Hochstrasser 2008); however, recently it has become called CRL4Cdt2 has been characterized, which couples clear that E2s can also contribute to processivity and proteolysis to DNA synthesis via an unusual mechanism specificity for ubiquitin chain nucleation and elongation that involves display of substrate degrons on the DNA (Jin et al. 2008; Ye and Rape 2009; Rodrigo-Brenni et al. polymerase processivity factor PCNA. Through its de- 2010; Saha et al. 2011; Wickliffe et al. 2011). Generally, struction of Cdt1, p21, and Set8, CRL4Cdt2 has emerged as the E3–substrate interaction involves the binding of a master regulator that prevents rereplication in S phase. -

SNP in Human ARHGEF3 Promoter Is Associated with Dnase
University of Groningen SNP in human ARHGEF3 promoter is associated with DNase hypersensitivity, transcript level and platelet function, and Arhgef3 KO mice have increased mean platelet volume Zou, Siying; Teixeira, Alexandra M.; Kostadima, Myrto; Astle, William J.; Radhakrishnan, Aparna; Simon, Lukas Mikolaj; Truman, Lucy; Fang, Jennifer S.; Hwa, John; Zhang, Ping-xia Published in: PLoS ONE DOI: 10.1371/journal.pone.0178095 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2017 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Zou, S., Teixeira, A. M., Kostadima, M., Astle, W. J., Radhakrishnan, A., Simon, L. M., ... Krause, D. S. (2017). SNP in human ARHGEF3 promoter is associated with DNase hypersensitivity, transcript level and platelet function, and Arhgef3 KO mice have increased mean platelet volume. PLoS ONE, 12(5), [e0178095]. https://doi.org/10.1371/journal.pone.0178095 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. -

Novel Pharmacological Maps of Protein Lysine Methyltransferases: Key for Target Deorphanization Obdulia Rabal* , Andrea Castellar and Julen Oyarzabal*
Rabal et al. J Cheminform (2018) 10:32 https://doi.org/10.1186/s13321-018-0288-5 RESEARCH ARTICLE Open Access Novel pharmacological maps of protein lysine methyltransferases: key for target deorphanization Obdulia Rabal* , Andrea Castellar and Julen Oyarzabal* Abstract Epigenetic therapies are being investigated for the treatment of cancer, cognitive disorders, metabolic alterations and autoinmune diseases. Among the diferent epigenetic target families, protein lysine methyltransferases (PKMTs), are especially interesting because it is believed that their inhibition may be highly specifc at the functional level. Despite its relevance, there are currently known inhibitors against only 10 out of the 50 SET-domain containing members of the PKMT family. Accordingly, the identifcation of chemical probes for the validation of the therapeutic impact of epigenetic modulation is key. Moreover, little is known about the mechanisms that dictate their substrate specifc- ity and ligand selectivity. Consequently, it is desirable to explore novel methods to characterize the pharmacologi- cal similarity of PKMTs, going beyond classical phylogenetic relationships. Such characterization would enable the prediction of ligand of-target efects caused by lack of ligand selectivity and the repurposing of known compounds against alternative targets. This is particularly relevant in the case of orphan targets with unreported inhibitors. Here, we frst perform a systematic study of binding modes of cofactor and substrate bound ligands with all available SET domain-containing PKMTs. Protein ligand interaction fngerprints were applied to identify conserved hot spots and contact-specifc residues across subfamilies at each binding site; a relevant analysis for guiding the design of novel, selective compounds. Then, a recently described methodology (GPCR-CoINPocket) that incorporates ligand contact information into classical alignment-based comparisons was applied to the entire family of 50 SET-containing proteins to devise pharmacological similarities between them.