Effective Strategies Used to Describe and Address the Burden of Sickle Cell Disease in The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Kyankwanzi Survey Report 2017
GROUND SURVEY FOR MEDIUM - LARGE MAMMALS IN KYANKWANZI CONCESSION AREA Report by F. E. Kisame, F. Wanyama, G. Basuta, I. Bwire and A. Rwetsiba, ECOLOGICAL MONITORING AND RESEARCH UNIT UGANDA WILDLIFE AUTHORITY 2018 1 | P a g e Contents Summary.........................................................................................................................4 1.0. INTRODUCTION ..................................................................................................5 1.1. Survey Objectives.....................................................................................................6 2.0. DESCRIPTION OF THE SURVEY AREA ..........................................................6 2.2. Location and Size .....................................................................................................7 2.2. Climate.....................................................................................................................7 2.3 Relief and Vegetation ................................................................................................8 3.0. METHOD AND MATERIALS..............................................................................9 Plate 1. Team leader and GPS person recording observations in the field.........................9 3.1. Survey design .........................................................................................................10 4.0. RESULTS .............................................................................................................10 4.1. Fauna......................................................................................................................10 -
Action for Rural Women's Empowerment
ACTION FOR RURAL WOMEN’S EMPOWERMENT Action For Rural ARUWE Women’s Empowerment Empowering Communities through women and Children (ARUWE) UGANDA ANNUAL REPORT 2014 - 2015 ANNUAL REPORT 1 ARUWE EMPOWERS THE RURAL COMMUNITIES THROUGH WOMEN AND CHILDREN 2015 Action For Rural ARUWE Women’s Empowerment Empowering Communities through women and Children ARUWE EMPOWERS THE RURAL COMMUNITIES THROUGH WOMEN AND CHILDREN ARUWE ANNUAL REPORT 2014 / 2015 ANNUAL REPORT 2 2015 Word From The Bod Chairperson________________________________________ 2 The Executive Director___________________________________________________ 3 Introduction_____________________________________________________________ 5 Vision, Mission, Core Programs__________________________________________ 5 Geographical Scope_____________________________________________________ 5 Promoting food security, nutrition and income generation________________ 8 Food security____________________________________________________________ 7 Integrating crop growing and livestock rearing___________________________ 9 Promotion of proper nutrition practices__________________________________ 9 Income generation______________________________________________________ 11 Community Fund - MFI__________________________________________________ 11 TABLE OF CONTENTS TABLE Rights awareness and leadership education_____________________________ 13 Water, Sanitation and Hygiene promotion________________________________ 14 Promotion of best practices_____________________________________________ 15 Community participation________________________________________________ -

HIV/AIDS Treatment and Care in a Long-Term Conflict Setting: Observations from the AIDS Support Organization (TASO) in the Teso Region Emma Smith SIT Study Abroad
SIT Graduate Institute/SIT Study Abroad SIT Digital Collections Independent Study Project (ISP) Collection SIT Study Abroad Spring 2008 HIV/AIDS Treatment and Care in a Long-Term Conflict Setting: Observations From The AIDS Support Organization (TASO) in the Teso Region Emma Smith SIT Study Abroad Follow this and additional works at: https://digitalcollections.sit.edu/isp_collection Recommended Citation Smith, Emma, "HIV/AIDS Treatment and Care in a Long-Term Conflict Setting: Observations From The AIDS Support Organization (TASO) in the Teso Region" (2008). Independent Study Project (ISP) Collection. 99. https://digitalcollections.sit.edu/isp_collection/99 This Unpublished Paper is brought to you for free and open access by the SIT Study Abroad at SIT Digital Collections. It has been accepted for inclusion in Independent Study Project (ISP) Collection by an authorized administrator of SIT Digital Collections. For more information, please contact [email protected]. HIV/AIDS Treatment and Care in a Long-Term Conflict Setting: Observations from The AIDS Support Organization (TASO) in the Teso Region Emma Smith Advisor: Alutia Samuel Academic Directors: Charlotte Mafumbo and Martha Wandera Location: TASO Soroti SIT Uganda Spring 2008 Dedication To all the people living with HIV/AIDS in Teso, who continue to live strongly despite decades of suffering from continuous war, displacement and neglect. May the world come to recognize the struggles that you live with. Acknowledgements There are so many people to whom thanks is owed, it would not be possible to acknowledge them all even if time and space allowed. Primarily, I would like to thank the clients of TASO Soroti, who so willingly welcomed a stranger into their communities and allowed so many questions to be asked of them. -

BETTER GROWTH, BETTER CITIES Achieving Uganda’S Development Ambition
BETTER GROWTH, BETTER CITIES Achieving Uganda’s Development Ambition A paper by the Government of Uganda and the New Climate Economy Partnership November 2016 THE REPUBLIC OF UGANDA THE REPUBLIC OF UGANDA About this paper The analysis in this paper was produced for the New Climate Partnership in Uganda research project, culminating in the report, Achieving Uganda’s Development Ambition: The Economic Impact of Green Growth – An Agenda for Action. This National Urban Transition paper is published as a supporting working paper and provides a fuller elaboration of the urbanisation elements in the broader report. Partners Achieving Uganda’s Development Ambition: The Economic Impact of Green Growth – An Agenda for Action was jointly prepared by the Government of Uganda through the Ministry of Finance, Planning and Economic Development (MFPED), the Ugandan Economic Policy Research Centre (EPRC) Uganda, the Global Green Growth Institute (GGGI), the New Climate Economy (NCE), and the Coalition for Urban Transitions (an NCE Special Initiative). Ministry of Finance, Planning and Economic Development Plot 2/12 Apollo Kaggwa Road P.O.Box 8147 Kampala, Uganda +256-414-707000 COALITION FOR URBAN TRANSITIONS A New Climate Economy Special Initiative Acknowledgements The project team members were Russell Bishop, Nick Godfrey, Annie Lefebure, Filippo Rodriguez and Rachel Waddell (NCE); Madina Guloba (EPRC); Maris Wanyera, Albert Musisi and Andrew Masaba (MPFED); and Samson Akankiza, Jahan-zeb Chowdhury, Peter Okubal and John Walugembe (GGGI). The technical -

Mapping Regional Reconciliation in Northern Uganda
Mapping Regional Reconciliation in Northern Uganda: A Case Study of the Acholi and Lango Sub-Regions Shilpi Shabdita Okwir Isaac Odiya Mapping Regional Reconciliation in Northern Uganda © 2015, Justice and Reconciliation Project, Gulu, Uganda All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, mechanical, photocopying, recording, or otherwise, without the prior written permission of the publisher. Applications for permission to reproduce or translate all or any part of this publication should be made to: Justice and Reconciliation Project Plot 50 Lower Churchill Drive, Laroo Division P.O. Box 1216 Gulu, Uganda, East Africa [email protected] Layout by Lindsay McClain Opiyo Front cover photo by Shilpi Shabdita Printed by the Justice and Reconciliation Project, Gulu, Uganda This publication was supported by a grant from USAID SAFE Program. However, the opinions and viewpoints in the report is not that of USAID SAFE Program. ii Justice and Reconciliation Project Acknowledgements This report was made possible with a grant from the United States Agency for International Development (USAID) Supporting Access to Justice, Fostering Equity and Peace (SAFE) Program for the initiation of the year-long project titled, “Across Ethnic Boundaries: Promoting Regional Reconciliation in Acholi and Lango Sub-Regions,” for which the Justice and Reconciliation Project (JRP) gratefully acknowledges their support. We are deeply indebted to Boniface Ojok, Head of Office at JRP, for his inspirational leadership and sustained guidance in this initiative. Special thanks to the enumerators Abalo Joyce, Acan Grace, Nyeko Simon, Ojimo Tycoon, Akello Paska Oryema and Adur Patritia Julu for working tirelessly to administer the opinion survey and to collect data, which has formed the blueprint of this report. -

Otuke District Local Government
CALL TO ACTION THE REPUBLIC OF UGANDA NUTRITION CHALLENGES/ GAPS CALL FOR ACTION RESPONSIBLE Otuke District Nutrition coordination committee Otuke was also supported to conduct a Food GOVERNANCE AREA OFFICE (DNCC), seven (7) Sub counties and One Security and Nutrition Assessments (FSNA). OTUKE DISTRICT LOCAL GOVERNMENT Coordination and Weak coordination mechanisms of Partner mapping required to know who DNFP, CAO Town council trained on multi sectoral nutrition FSNA data was not available previously partnerships: nutrition actions at all levels. is where and doing what. DNCC/SNCC ADVOCACY BRIEF ON STRENGTHENING NUTRITION GOVERNANCE FOR MULTI-SECTORAL RESPONSE implementation for improved nutrition unavailable therefore this first FSNA data will members need to be oriented on their outcomes. be used as a baseline to compare progress roles and responsibilities in achievement of health, nutrition and WASH The district conducted quarterly DNCC meetings Establish joint planning and strategic indicators in subsequent FSNAs. Annual FSNAs and support supervision activities aimed at coordination mechanisms amongst will be conducted to assess annual progress. strengthening the accountability framework for partners in the district to reduce on Multisectoral nutrition actions implemented in The Otuke DNCC has been trained on nutrition duplication of resources and achieve sustainable results Otuke district. governance and supported to use reporting templates and monitoring tools previously Systems capacity Lack of clarity on nutrition sensitive Orientation -

A Monthly Newsletter on Food Security and Vulnerability in Uganda
A Monthly Newsletter on Food Security and Vulnerability in Uganda Number 02/2001 15 February, 2001 Summary The NGO, Concern Worldwide, reports that households in Katakwi District (eastern Uganda) are experiencing moderate food insecurity. The most affected people are located in Kapelebyong and Usuk Counties on the border with Moroto District, where there is limited access to food and malnutrition rates are high. Only households in Magoro Sub-County of Usuk, received about 100 kg of maize and beans from another NGO, Hands on Service, in early December. No further assistance has been provided and there are no immediate plans to provide additional food aid to the affected people in the district. Because of the expected increase in demand on meager food supplies and other resources, many residents are worried about the Karimojong pastoralists and their normal dry season migratory pattern into Katakwi District in search of water and pastures for their livestock. Large livestock herds may cause destruction of crops and vegetation, increasing vulnerability to food insecurity and competition for already diminishing pastures. District authorities and local residents also are concerned about the possibility of increasing tensions and civil insecurity due to the presence of armed pastoralists. To ensure peace and security in the district, the Government has increased deployment of Uganda People’s Defence Force personnel. Concern Worldwide affirms that even though the population may require assistance after February 2001 when household food stocks run low, it is imperative for adequate civil security to be maintained to allow proper identification and targeting of the most affected households before carrying out any mitigation program for maximum benefit. -

WHO UGANDA BULLETIN February 2016 Ehealth MONTHLY BULLETIN
WHO UGANDA BULLETIN February 2016 eHEALTH MONTHLY BULLETIN Welcome to this 1st issue of the eHealth Bulletin, a production 2015 of the WHO Country Office. Disease October November December This monthly bulletin is intended to bridge the gap between the Cholera existing weekly and quarterly bulletins; focus on a one or two disease/event that featured prominently in a given month; pro- Typhoid fever mote data utilization and information sharing. Malaria This issue focuses on cholera, typhoid and malaria during the Source: Health Facility Outpatient Monthly Reports, Month of December 2015. Completeness of monthly reporting DHIS2, MoH for December 2015 was above 90% across all the four regions. Typhoid fever Distribution of Typhoid Fever During the month of December 2015, typhoid cases were reported by nearly all districts. Central region reported the highest number, with Kampala, Wakiso, Mubende and Luweero contributing to the bulk of these numbers. In the north, high numbers were reported by Gulu, Arua and Koti- do. Cholera Outbreaks of cholera were also reported by several districts, across the country. 1 Visit our website www.whouganda.org and follow us on World Health Organization, Uganda @WHOUganda WHO UGANDA eHEALTH BULLETIN February 2016 Typhoid District Cholera Kisoro District 12 Fever Kitgum District 4 169 Abim District 43 Koboko District 26 Adjumani District 5 Kole District Agago District 26 85 Kotido District 347 Alebtong District 1 Kumi District 6 502 Amolatar District 58 Kween District 45 Amudat District 11 Kyankwanzi District -

Sustainable and Resilient Grandmothers Project Report in Mulagi, Kyankwanzi District, Uganda
SUSTAINABLE AND RESILIENT GRANDMOTHERS PROJECT REPORT IN MULAGI, KYANKWANZI DISTRICT, UGANDA End of project report from 1st February 2013 to 31st July 2013 Funded by The Stephen Lewis Foundation Agency Agreement Date: 1stAugust 2012 Agency Agreement Amount: 25,000 CN Compiled and Prepared by: Agnes Mirembe Sylvia Nalubega Action For Rural ARUWE Women’s Empowerment SUSTAINABLETimothy Gasana AND RESILIENT GRANDMOTHERS PROJECT REPORT 1 Peace Rwankiiko INTRODUCTION ince 2010 Stephen Lewis Foundation has partnered with Action for Rural Women’s Empowerment (ARUWE) to support grandmothers and their Orphaned and Vulnerable Children in Mulagi Sub County, Kyankwanzi district. InS 2012-2013 SLF supported the “Sustainable and Resilient Grandmothers Project in Mulagi Sub County, Kyankwanzi district” as a continuation of the last project phase. The main goal of the project was to strengthen the capacities of 150 Grandmothers and 1000 orphaned grandchildren to learn new skills; increase household food security; earn, manage and reinvest income; serve as moral leaders to their families and communities; and keep their orphans safe, healthy, educated and loved. In order to achieve the main project goal, the objectives set included; 1. Increase household food security and income through increased skills, access to agricultural inputs and revolving loan 2. Support health, nutrition and psycho-social enhancing interventions to 150 Grandmothers of orphaned grandchildren in Mulagi Sub County, Kyankwanzi district. Objective One: To increase household food security and income through increased skills, access to agricultural inputs and revolving loan, the project implemented the following activities; Training grandmothers and their caretakers in sustainable agricultural methods To increase household food production, the project conducted trainings in sustainable agriculture methods and practices among the 150 grandmothers and their caretakers 100. -
Tilenga & Eacop Projects with a Socio-Economic Interest for Uganda
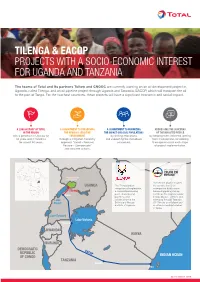
TILENGA & EACOP PROJECTS WITH A SOCIO-ECONOMIC INTEREST FOR UGANDA AND TANZANIA The teams of Total and its partners Tullow and CNOOC are currently working on an oil development project in Uganda, called Tilenga, and an oil pipeline project through Uganda and Tanzania, EACOP, which will transport the oil to the port of Tanga. For the two host countries, these projects will have a significant economic and social impact. A LONG HISTORY OF TOTAL A COMMITMENT TO PRESERVING A COMMITMENT TO MINIMIZING ADDRESSING THE CONCERNS IN THE REGION THE REGION'S SENSITIVE THE IMPACT ON LOCAL POPULATIONS OF THE IMPACTED PEOPLE with a presence in Uganda for ENVIRONMENT by limiting relocations by keeping them informed, getting 60 years and in Tanzania through a mitigation hierarchy and supporting the individuals them involved and considering for almost 50 years. approach “Avoid – Reduce/ concerned. their opinions into each stage Restore – Compensate” of project implementation. and concrete actions. SOUDAN DU SUD ÉTHIOPIE Murchison Falls National Park The EACOP project involves UGANDA The Tilenga project the construction of an comprises oil exploration, underground hydrocarbon Tilenga a crude oil processing transport pipeline starting Hoima plant, underground just inside the Uganda border pipelines, and (Hoima District - 297km) and Lake infrastructure in the extending through Tanzania Albert Buliisa and Nwoya (1147km) to an oil depot and districts of Uganda. an offshore loading terminal in Tanga. Lake Edward Lake Victoria Bukoba RWANDA KENYA BURUNDI DEMOCRATIC EACOP REPUBLIC INDIAN OCEAN OF CONGO TANZANIA Singida Tanga SEPTEMBER 2019 ZAMBIE MOZAMBIQUE MALAWI SOUDAN DU SUD THIOPIE FOCUS ON THE TILENGA PROJECT Total E&P Uganda, fully aware of the project's sensitive nature, has placed particular emphasis on environmental and societal issues, with a specific commitment to leaving the site in a better state than it was before the work started and to limiting residents' relocations as much as possible. -

Funding Going To
% Funding going to Funding Country Name KP‐led Timeline Partner Name Sub‐awardees SNU1 PSNU MER Structural Interventions Allocated Organizations HTS_TST Quarterly stigma & discrimination HTS_TST_NEG meetings; free mental services to HTS_TST_POS KP clients; access to legal services PrEP_CURR for KP PLHIV PrEP_ELIGIBLE Centro de Orientacion e PrEP_NEW Dominican Republic $ 1,000,000.00 88.4% MOSCTHA, Esperanza y Caridad, MODEMU Region 0 Distrito Nacional Investigacion Integral (COIN) PrEP_SCREEN TX_CURR TX_NEW TX_PVLS (D) TX_PVLS (N) TX_RTT Gonaives HTS_TST KP sensitization focusing on Artibonite Saint‐Marc HTS_TST_NEG stigma & discrimination, Nord Cap‐Haitien HTS_TST_POS understanding sexual orientation Croix‐des‐Bouquets KP_PREV & gender identity, and building Leogane PrEP_CURR clinical providers' competency to PrEP_CURR_VERIFY serve KP FY19Q4‐ KOURAJ, ACESH, AJCCDS, ANAPFEH, APLCH, CHAAPES, PrEP_ELIGIBLE Haiti $ 1,000,000.00 83.2% FOSREF FY21Q2 HERITAGE, ORAH, UPLCDS PrEP_NEW Ouest PrEP_NEW_VERIFY Port‐au‐Prince PrEP_SCREEN TX_CURR TX_CURR_VERIFY TX_NEW TX_NEW_VERIFY Bomu Hospital Affiliated Sites Mombasa County Mombasa County not specified HTS_TST Kitui County Kitui County HTS_TST_NEG CHS Naishi Machakos County Machakos County HTS_TST_POS Makueni County Makueni County KP_PREV CHS Tegemeza Plus Muranga County Muranga County PrEP_CURR EGPAF Timiza Homa Bay County Homa Bay County PrEP_CURR_VERIFY Embu County Embu County PrEP_ELIGIBLE Kirinyaga County Kirinyaga County HWWK Nairobi Eastern PrEP_NEW Tharaka Nithi County Tharaka Nithi County -

Dairy Development in Uganda
DAIRY DEVELOPMENT IN UGANDA MinistryThe Republic of Agriculture of Uganda Animal Industry and Fisheries (MAAIF) Food and Agriculture A Review of Uganda’s Dairy Industry Organization of the United Nations Dairy Development Authority (DDA) Author: David Balikowa National Consultant, GOU/FAO Dairy Project, TCP/UGA/3202(D); Senior Business Advisor, TechnoServe/East Africa Dairy Development Project (EADD) March 2011 i TABLE OF CONTENTS LIST OF TABLES .................................................................................................................................. v LIST OF FIGURES ................................................................................................................................ v ACRONYMS AND ABBREVIATIONS .....................................................................................................vi EXECUTIVE SUMMARY ...................................................................................................................... vii CHAPTER 1 ........................................................................................................................................ 1 1. INTRODUCTION ......................................................................................................................... 1 1.1 Geographical Location of Uganda ....................................................................................... 1 1.2 Contribution of Agriculture to the National Economy ......................................................... 1 1.3 Historical Overview of