Flecainide Increases Kir2.1 Currents by Interacting with Cysteine 311, Decreasing the Polyamine- Induced Rectification
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
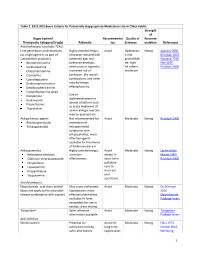
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -
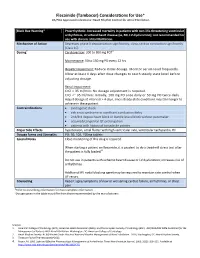
Flecainide Considerations For
Flecainide (Tambocor) Considerations for Use* US/FDA Approved Indications: Heart Rhythm Control for Atrial Fibrillation Black Box Warning* Proarrhythmic. Increased mortality in patients with non-life-threatening ventricular arrhythmias, structural heart disease (ie, MI, LV dysfunction); not recommended for use with chronic atrial fibrillation. Mechanism of Action Depresses phase 0 depolarization significantly, slows cardiac conduction significantly (Class 1C). Dosing† Cardioversion: 200 to 300 mg PO‡1 Maintenance: 50 to 150 mg PO every 12 hrs Hepatic Impairment: Reduce initial dosage. Monitor serum level frequently. Allow at least 4 days after dose changes to reach steady state level before adjusting dosage. Renal Impairment: CrCl > 35 ml/min: No dosage adjustment is required. CrCl <= 35 ml/min: Initially, 100 mg PO once daily or 50 mg PO twice daily. Adjust dosage at intervals > 4 days, since steady-state conditions may take longer to achieve in these patient Contraindications cardiogenic shock sick sinus syndrome or significant conduction delay 2nd/3rd degree heart block or bundle brand block without pacemaker acquired/congenital QT prolongation patients with history of torsade de pointes Major Side Effects hypotension, atrial flutter with high ventricular rate, ventricular tachycardia, HF Dosage forms and Strengths PO: 50, 100, 150mg tablets Special Notes Close monitoring of this drug is required. When starting a patient on flecainide, it is prudent to do a treadmill stress test after the patient is fully loaded.4 Do not use in patients with ischemic heart disease or LV dysfunction; increases risk of arrhythmias. Additional AV nodal blocking agent may be required to maintain rate control when AF recurs. -

A Proposal for the Clinical Use of Flecainide
A Proposal for the Clinical Use of Flecainide JEFFREY L. ANDERSON, MD, JAMES R. STEWART, MD, and BARRY J. CREVEY, MD Effective antiarrhythmic therapy requires a carefully sponse rate is observed in preventing induction of considered approach, including an understanding sustained ventricular tachycardia, and these pa- of the arrhythmia, the underlying cardiac disease tients should be carefully selected. Flecainide is and the drug’s pharmacokinetics. Flecainide is a promising in the treatment of supraventricular new antiarrhythmic drug that may soon be released tachycardias using atrioventricular nodal or extra- for general use. Flecainide demonstrates unsur- nodal reentrant pathways, although this use is still passed efficacy in chronic ventricular arrhythmias investigational in the United States. The drug’s use in stable patients and may become a first-choice for arrhythmias during acute myocardial infarction drug because of its ease of administration, efficacy requires further study. Flecainide possesses modest and favorable tolerance. Twice-daily dosing with negative inotropic potential. Proarrhythmic or other 100 to 200 mg usually provides effective therapy. adverse reactions have occurred primarily in set- Clinical experience suggests flecainide to be indi- tings of high drug level, poor ventricular function cated in the treatment of uniform and multiform or refractory, malignant arrhythmias, suggesting ventricular premature complexes, coupled ven- caution in these groups. tricular premature complexes, and episodes of nonsustained -

Guideline for Preoperative Medication Management
Guideline: Preoperative Medication Management Guideline for Preoperative Medication Management Purpose of Guideline: To provide guidance to physicians, advanced practice providers (APPs), pharmacists, and nurses regarding medication management in the preoperative setting. Background: Appropriate perioperative medication management is essential to ensure positive surgical outcomes and prevent medication misadventures.1 Results from a prospective analysis of 1,025 patients admitted to a general surgical unit concluded that patients on at least one medication for a chronic disease are 2.7 times more likely to experience surgical complications compared with those not taking any medications. As the aging population requires more medication use and the availability of various nonprescription medications continues to increase, so does the risk of polypharmacy and the need for perioperative medication guidance.2 There are no well-designed trials to support evidence-based recommendations for perioperative medication management; however, general principles and best practice approaches are available. General considerations for perioperative medication management include a thorough medication history, understanding of the medication pharmacokinetics and potential for withdrawal symptoms, understanding the risks associated with the surgical procedure and the risks of medication discontinuation based on the intended indication. Clinical judgement must be exercised, especially if medication pharmacokinetics are not predictable or there are significant risks associated with inappropriate medication withdrawal (eg, tolerance) or continuation (eg, postsurgical infection).2 Clinical Assessment: Prior to instructing the patient on preoperative medication management, completion of a thorough medication history is recommended – including all information on prescription medications, over-the-counter medications, “as needed” medications, vitamins, supplements, and herbal medications. Allergies should also be verified and documented. -

Drug Interaction of Amiodarone, Flecainide, Metoprolol and Diltiazem Leading to Heart Block Vijay Kahajuria, Sanjeev Gupta, Roshi, Neelam Rani
JK SCIENCE CASE REPORT Drug Interaction of Amiodarone, Flecainide, Metoprolol and Diltiazem leading to Heart Block Vijay Kahajuria, Sanjeev Gupta, Roshi, Neelam Rani Abstract Amiodarone, flecainide, metoprolol and diltiazem individually are known to cause heart blocks due to their cardiac depressant property but the current case report is worth reporting because it resulted because of drug interaction of multiple drugs due to possible medication error. Key Words Amiodarone, Flecainide, Metoprolol, Diltiazem, Heart Blocks Introduction Adverse Drug Reactions (ADRs) in cardiology are thyroid dysfunction. His hemoglobin levels were 13g/dl, common. There are numerous reports of drug induced total leucocyte count 6000/UL, neutrophils 56.5%, bradycardia and heart block (1). Co-morbid conditions lymphocytes 30.5%, eosinophils 5.2%, monocytes often coexist with cardiac ailments and results in 7.6%,basophils 0.2%, platelet count 1.5 lacs, serum urea- polypharmacy which enhance the chances of drug 33mg/dl, serum creatinine 0.90mg/dl, prothrombin time interactions culminating to adverse events. In the current 10.9 sec ,INR 1.04,hepatitis serology was non reactive, study report patient was prescribed multiple drugs for blood sugar random 182 mg/dl, TSH 1.4uIU/ml, serum cardiac arrhythmia and he presented with heart block sodium 142 mmol/L,serum potassium 5 mmol/L. and bradycardia. Though amiodarone, flecainide, He was prescribed tab.atenolol 20mg and metoprolol and diltiazem individually are known to cause tab.procainamide 20mg which did not bring any relief. heart blocks due to their cardiac depressant property but So radiofrequency ablation was done which was the current case report is worth reporting because it uneventful. -

Flecainide in Ventricular Arrhythmias: from Old Myths to New Perspectives
Journal of Clinical Medicine Review Flecainide in Ventricular Arrhythmias: From Old Myths to New Perspectives Carlo Lavalle 1,*, Sara Trivigno 1 , Giampaolo Vetta 1 , Michele Magnocavallo 1 , Marco Valerio Mariani 1 , Luca Santini 2, Giovanni Battista Forleo 3 , Massimo Grimaldi 4, Roberto Badagliacca 1, Luigi Lanata 5 and Renato Pietro Ricci 6 1 Department of Cardiovascular, Respiratory, Nephrological, Anesthesiological and Geriatric Sciences, Sapienza University of Rome, Policlinico Umberto I, 00161 Rome, Italy; [email protected] (S.T.); [email protected] (G.V.); [email protected] (M.M.); [email protected] (M.V.M.); [email protected] (R.B.) 2 Department of Cardiology, Ospedale GB Grassi, 00121 Ostia, Italy; [email protected] 3 Department of Cardiology, Azienda Ospedaliera-Universitaria “Luigi Sacco”, 20057 Milan, Italy; [email protected] 4 Department of Cardiology, Ospedale Generale Regionale F. Miulli, Acquaviva delle Fonti, 70021 Bari, Italy; fi[email protected] 5 Medical Affairs Department, Dompé Farmaceutici SpA, 20057 Milan, Italy; [email protected] 6 Centro Cardio-Aritmologico, 00152 Rome, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-335376901 Abstract: Flecainide is an IC antiarrhythmic drug (AAD) that received in 1984 Food and Drug Administration approval for the treatment of sustained ventricular tachycardia (VT) and subsequently Citation: Lavalle, C.; Trivigno, S.; for rhythm control of atrial fibrillation (AF). Currently, flecainide is mainly employed for sinus Vetta, G.; Magnocavallo, M.; Mariani, rhythm maintenance in AF and the treatment of idiopathic ventricular arrhythmias (IVA) in absence M.V.; Santini, L.; Forleo, G.B.; Grimaldi, M.; Badagliacca, R.; Lanata, of ischaemic and structural heart disease on the basis of CAST data. -

FLECAINIDE ACETATE Tablets, USP Rx Only DESCRIPTION CLINICAL PHARMACOLOGY
FLECAINIDE ACETATE Tablets, USP Rx only DESCRIPTION Flecainide is an antiarrhythmic drug available in tablets of 50, 100, or 150 mg for oral administration. Flecainide acetate is benzamide, N-(2-piperidinylmethyl)-2,5-bis (2,2,2-trifluoroethoxy)- monoacetate. The structural formula is given below. Flecainide acetate is a white crystalline substance with a pKa of 9.3. It has an aqueous solubility of 48.4 mg/mL at 37°C. Flecainide Acetate Tablets, USP also contain: croscarmellose sodium, hydrogenated vegetable oil, magnesium stearate, microcrystalline cellulose, and pregelatinized starch. CLINICAL PHARMACOLOGY Flecainide has local anesthetic activity and belongs to the membrane stabilizing (Class 1) group of antiarrhythmic agents; it has electrophysiologic effects characteristic of the IC class of antiarrhythmics. Electrophysiology In man, flecainide produces a dose-related decrease in intracardiac conduction in all parts of the heart with the greatest effect on the His-Purkinje system (H-V conduction). Effects upon atrioventricular (AV) nodal conduction time and intra-atrial conduction times, although present, are less pronounced than those on ventricular conduction velocity. Significant effects on refractory periods were observed only in the ventricle. Sinus node recovery times (corrected) following pacing and spontaneous cycle lengths are somewhat increased. This latter effect may become significant in patients with sinus node dysfunction. (See WARNINGS.) Flecainide causes a dose-related and plasma-level related decrease in single and multiple PVCs and can suppress recurrence of ventricular tachycardia. In limited studies of patients with a history of ventricular tachycardia, flecainide has been successful 30 to 40% of the time in fully suppressing the inducibility of arrhythmias by programmed electrical stimulation. -

Oral Antiarrhythmic Drugs in Converting Recent Onset Atrial Fibrillation
Review article Oral antiarrhythmic drugs in converting recent onset atrial fibrillation • Vera H.M. Deneer, Marieke B.I. Borgh, J. Herre Kingma, Loraine Lie-A-Huen and Jacobus R.B.J. Brouwers Introduction Pharm World Sci 2004; 26: 66–78. © 2004 Kluwer Academic Publishers. Printed in the Netherlands. Atrial fibrillation is the most common arrhythmia. The incidence of atrial fibrillation depends on the age of V.H.M. Deneer (correspondence, e-mail: the study population. The incidence varies between 2 [email protected]), M.B.I. Borgh, L. Lie-A-Huen: Department of Clinical Pharmacy or 3 new cases per 1,000 population per year between J.H. Kingma: Department of Cardiology, St Antonius Hospital, the ages of 55 and 64 years to 35 new cases per 1,000 Koekoekslaan 1, 3435 CM Nieuwegein, The Netherlands population per year between the ages of 85 and 94 J.R.B.J. Brouwers: Groningen University Institute for Drug 1 Exploration (GUIDE), Section of Pharmacotherapy, University years . Treatment of an episode of paroxysmal atrial fi- of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, brillation consists of restoring sinus rhythm by DC- The Netherlands electrical cardioversion or by the intravenous adminis- Key words tration of an antiarrhythmic drug, but frequently the Atrial fibrillation arrhythmia spontaneously terminates1–3. After one or Amiodarone more episodes of atrial fibrillation chronic prophylac- Antiarrhythmic drugs Digoxin tic treatment with an antiarrhythmic drug is often Episodic treatment started for maintenance of sinus rhythm4–8. Another Flecainide treatment strategy consists of allowing the arrhythmia Propafenone Quinidine to exist in combination with pharmacological ven- Sotalol tricular rate control. -

2020 Aetna Standard Plan
Plan for your best health Aetna Standard Plan Aetna.com Aetna is the brand name used for products and services provided by one or more of the Aetna group of subsidiary companies, including Aetna Life Insurance Company and its affiliates (Aetna). Aetna Pharmacy Management refers to an internal business unit of Aetna Health Management, LLC. Aetna Pharmacy Management administers, but does not offer, insure or otherwise underwrite the prescription drug benefits portion of your health plan and has no financial responsibility therefor. 2020 Pharmacy Drug Guide - Aetna Standard Plan Table of Contents INFORMATIONAL SECTION..................................................................................................................6 *ADHD/ANTI-NARCOLEPSY/ANTI-OBESITY/ANOREXIANTS* - DRUGS FOR THE NERVOUS SYSTEM.................................................................................................................................16 *ALLERGENIC EXTRACTS/BIOLOGICALS MISC* - BIOLOGICAL AGENTS...............................18 *ALTERNATIVE MEDICINES* - VITAMINS AND MINERALS....................................................... 19 *AMEBICIDES* - DRUGS FOR INFECTIONS.....................................................................................19 *AMINOGLYCOSIDES* - DRUGS FOR INFECTIONS.......................................................................19 *ANALGESICS - ANTI-INFLAMMATORY* - DRUGS FOR PAIN AND FEVER............................19 *ANALGESICS - NONNARCOTIC* - DRUGS FOR PAIN AND FEVER......................................... -

Class I Antiarrhythmics Did You Know?
February 2018 Poison Center Hotline: 1-800-222-1222 The Maryland Poison Center’s Monthly Update: News, Advances, Information Class I Antiarrhythmics The Maryland Poison Center was consulted about a 4 year old male who present- ed to the emergency department with persistent junctional reciprocating tachy- cardia after ingestion of up to 2,000 mg of his own flecainide. On arrival, he was hypotensive and in a wide complex tachycardia on EKG. He was cardioverted 3 times, which resulted in transient stabilization of heart rhythm, but with a return to ventricular tachycardia. Eventually he stabilized after multiple doses of sodium bicarbonate. EKG showed a QRS of 158 msec and a QTc of 700 msec. He contin- ued to have recurrent arrhythmias for greater than 24 hours after his ingestion, but recovered completely by 30 hours post ingestion. Did you know? I trained in the post CAST-1/2 and AFFIRM era, when we learned “rate control good, rhythm control bad.” When it comes to overdoses, the mantra is more ap- Lipid emulsion may clog propriately “rate control bad, rhythm control bad.” Class I antiarrhythmics are extracorporeal membrane incredibly toxic in overdose. These drugs include disopyramide, quinidine, pro- oxygen machines. cainamide, mexiletine, lidocaine, flecainide, and propafenone. All of them are sodium channel blockers, but are subdivided into class Ia, Ib, and Ic based on Extracorporeal membrane oxygen their rate of interaction with the sodium channel. Toxicity is dose dependent, but (ECMO) is becoming more popular in significant toxicity has been reported with inadvertent double doses of flecainide the toxicology world as an assist (Am J Emerg Med 2012;30:2095.e1-2). -

Neemmc Guidelines for Tablet Crushing and Administration Via Enteral Feeding Tubes
NEEMMC GUIDELINES FOR TABLET CRUSHING AND ADMINISTRATION VIA ENTERAL FEEDING TUBES KEY TO DRUG ADMINISTRATION GUIDELINES Please follow the guidelines in order, as shown in the chart (i.e. number 1 is the first choice of which form to administer the drug in). A Tablet will disperse in 1-2 minutes. B Tablet will disperse in greater than 2 minutes. C Liquid preparation available. D Dilute reconstituted injection with 30-60ml of water before administering. E Oral solution/suspension can be prepared by local pharmacy or Non Sterile Preparative Services (PSU at Colchester General Hospital). Note: It is an unlicensed use to crush tablets, open capsules and make extemporaneous suspensions. However, using tablets within these guidelines is covered by the Trust for legal/vicarious liability. For more information on the administration via different tubes, please contact Medicines Information on ext. 2161. Drug Key code Information ACETAZOLAMIDE 1. A* * Diamox SR capsules can be opened and 2. D the contents flushed down the enteral tube 3. E ACICLOVIR 1. C * Dispersible tablets available 2. A* ALFACALCIDOL C* * Drops available. Sensitive to light ALFUZOSIN A Beware of sudden hypotensive effect if giving crushed tablets. Monitor BP and ensure patient is lying down prior to administering the dose. Do not crush slow release preparations. ALLOPURINOL 1. B 2. E ALVERINE Content of capsules is very bitter, and might numb the tongue and throat. AMILORIDE 1. B 2. C* * Liquid available. AMINOPHYLLINE Convert to theophylline liquid equivalent. Do not crush SR preparations. AMIODARONE B AMITRIPTYLINE 1. C 2. B AMLODIPINE A A Tablet will disperse in 1-2 minutes. -

Medications to Avoid in Long QT Syndrome
Jackie Crawford Cardiac Inherited Disease Co-ordinator C/- Paediatric Cardiology; Level 3 Starship; Auckland City Hospital Private Bag 92024; Auckland Cardiac Inherited Disease Registry Phone: (09) 3074949 ext 23634 www.cidg.org Fax: (09) 6309877 Email: [email protected] Medications to be avoided, or requiring special caution, in people with Long QT syndrome This list includes medications which prolong the QT interval and is meant as a guide for people with Long QT syndrome, or acquired long QT interval from heart muscle disease, and their parents or guardians. It should not be seen as all inclusive. Those prescribing any medication to someone with Long QT syndrome should always check the drug specifications and contra-indications. The list has been compiled by review of publications and/or drug advice sheets provided with medications. Check also www.SADS.org and www.Torsades .org. Antibiotics Erythromycin, Clarithromycin, Gatifloxacin, levofloxacin, Moxifloxacin Sulfamethoxazole-trimethoprim (Septrin/Bactrim), Spiramycin, Pentamidine Antihistamines Terfenadine, Astemizole, Diphenhydramine, (These are particularly to be avoided (even in normal subjects) in combination with Erythromycin or grapefruit juice or the antifungals ketoconazole, miconazole, fluconazole or itraconazole) [Antihistamines that may be used safely are loratidine, cetirizine and fexofenadine, and phenergan] Appetite suppressants Fenfluramine, phentermine, Sibutramine Asthma treatments The Beta-2 agonists (e.g. Terbutaline, Salbutamol, Salmeterol) both work against