Genome-Wide Copy Number Variation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Proquest Dissertations
nm u Ottawa L'Universite canadienne Canada's university run FACULTE DES ETUDES SUPERIEURES ^=1 FACULTY OF GRADUATE AND ET POSTOCTORALES U Ottawa POSDOCTORAL STUDIES L'Univeisittf canathenne Canada's university Anna Francina Jackson „_„^^.^^_^^^.„^ M.Sc. (Cellular and Molecular Medicine) __„.___._ Department of Cellular and Molecular Medicine The Functional Characterization of Wdr68 Regulation of Pax7 Activity in Myogenesis TITRE DE LA THESE / TITLE OF THESIS M. Rudnicki „„„„_.___ N;LWij3CT:Berg©ron RJjcreaton_ Gary W. Slater Le Doyen de la Faculte des etudes superieures et postdoctorales / Dean of the Faculty of Graduate and Postdoctoral Studies The Functional Characterization of Wdr68 Regulation of Pax7 Activity in Myogenesis Anna Francina Jackson This thesis is submitted to the Faculty of Graduate and Postdoctoral Studies in partial fulfillment of the requirements for a Master of Science degree in Cellular and Molecular Medicine. Department of Cellular and Molecular Medicine Faculty of Medicine University of Ottawa May 14, 2010 ©Anna Francina Jackson, Ottawa, Canada, 2010 Library and Archives Bibliotheque et 1*1 Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington OttawaONK1A0N4 OttawaONK1A0N4 Canada Canada Your file Votre reference ISBN: 978-0-494-74221-1 Our file Notre reference ISBN: 978-0-494-74221-1 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library and permettant a la Bibliotheque -

DYRK1A Binds to an Evolutionarily Conserved WD40-Repeat Title Protein WDR68 and Induces Its Nuclear Translocation
DYRK1A binds to an evolutionarily conserved WD40-repeat Title protein WDR68 and induces its nuclear translocation. Author(s) Miyata, Yoshihiko; Nishida, Eisuke Citation Biochimica et biophysica acta (2011), 1813(10): 1728-1739 Issue Date 2011-10 URL http://hdl.handle.net/2433/148020 © 2011 Elsevier B.V.; This is not the published version. Please cite only the published version.; この論文は出版社版であり Right ません。引用の際には出版社版をご確認ご利用ください 。 Type Journal Article Textversion author Kyoto University *REVISED Manuscript (text UNmarked) Click here to view linked References DYRK1A binds to an evolutionarily conserved WD40-repeat protein WDR68 and induces its nuclear translocation Yoshihiko Miyata*, Eisuke Nishida Department of Cell and Developmental Biology, Graduate School of Biostudies, Kyoto University, Kitashirakawa Oiwake-cho, Kyoto 606-8502, Japan * Corresponding author. Department of Cell & Developmental Biology, Graduate School of Biostudies, Kyoto University, Kitashirakawa Oiwake-cho, Sakyo-ku, Kyoto 606-8502, Japan. Tel.: +81-75-753-4231; fax: +81-75-753-4235. 1 ABSTRACT DYRK1A is encoded in the Down’s syndrome critical region on human chromosome 21, and plays an important role in the functional and developmental regulation of many types of cells, including neuronal cells. Here we have identified WDR68, an evolutionarily conserved protein with WD40-repeat domains, as a cellular binding partner of DYRK1A. WDR68 was originally identified in petunia as AN11 that controls the pigmentation of flowers by stimulating the transcription of anthocyanin biosynthetic genes. Experiments with RNA interference showed that WDR68 was indispensable for the optimal proliferation and survival of mammalian cultured cell, and WDR68 depletion induced cell apoptosis. DYRK1A and DYRK1B, but not DYRK2, DYRK3, or DYRK4, bound to endogenous and expressed WDR68. -

Estimation of Non-Null SNP Effect Size Distributions Enables the Detection
bioRxiv preprint doi: https://doi.org/10.1101/597484; this version posted May 13, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Estimation of Non-null SNP Effect Size Distributions Enables 2 the Detection of Enriched Genes Underlying Complex Traits 3 1,2 1,2 2-4 4 Wei Cheng , Sohini Ramachandran y, and Lorin Crawford y 5 1 Department of Ecology and Evolutionary Biology, Brown University, Providence, RI, 6 USA 7 2 Center for Computational Molecular Biology, Brown University, Providence, RI, USA 8 3 Department of Biostatistics, Brown University, Providence, RI, USA 9 4 Center for Statistical Sciences, Brown University, Providence, RI, USA 10 Corresponding E-mail: [email protected]; lorin [email protected] y 11 Abstract 12 Traditional univariate genome-wide association studies generate false positives and negatives due to 13 difficulties distinguishing associated variants from variants with spurious nonzero effects that do not 14 directly influence the trait. Recent efforts have been directed at identifying genes or signaling pathways 15 enriched for mutations in quantitative traits or case-control studies, but these can be computationally 16 costly and hampered by strict model assumptions. Here, we present gene-", a new approach for identifying 17 statistical associations between sets of variants and quantitative traits. Our key insight is that enrichment 18 studies on the gene-level are improved when we reformulate the genome-wide SNP-level null hypothesis 19 to identify spurious small-to-intermediate SNP effects and classify them as non-causal. -

Mammalian PRC1 Complexes: Compositional Complexity and Diverse Molecular Mechanisms
International Journal of Molecular Sciences Review Mammalian PRC1 Complexes: Compositional Complexity and Diverse Molecular Mechanisms Zhuangzhuang Geng 1 and Zhonghua Gao 1,2,3,* 1 Departments of Biochemistry and Molecular Biology, Penn State College of Medicine, Hershey, PA 17033, USA; [email protected] 2 Penn State Hershey Cancer Institute, Hershey, PA 17033, USA 3 The Stem Cell and Regenerative Biology Program, Penn State College of Medicine, Hershey, PA 17033, USA * Correspondence: [email protected] Received: 6 October 2020; Accepted: 5 November 2020; Published: 14 November 2020 Abstract: Polycomb group (PcG) proteins function as vital epigenetic regulators in various biological processes, including pluripotency, development, and carcinogenesis. PcG proteins form multicomponent complexes, and two major types of protein complexes have been identified in mammals to date, Polycomb Repressive Complexes 1 and 2 (PRC1 and PRC2). The PRC1 complexes are composed in a hierarchical manner in which the catalytic core, RING1A/B, exclusively interacts with one of six Polycomb group RING finger (PCGF) proteins. This association with specific PCGF proteins allows for PRC1 to be subdivided into six distinct groups, each with their own unique modes of action arising from the distinct set of associated proteins. Historically, PRC1 was considered to be a transcription repressor that deposited monoubiquitylation of histone H2A at lysine 119 (H2AK119ub1) and compacted local chromatin. More recently, there is increasing evidence that demonstrates the transcription activation role of PRC1. Moreover, studies on the higher-order chromatin structure have revealed a new function for PRC1 in mediating long-range interactions. This provides a different perspective regarding both the transcription activation and repression characteristics of PRC1. -

Recalibrating the Genome-Wide Association Null Hypothesis
bioRxiv preprint doi: https://doi.org/10.1101/597484; this version posted October 4, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Recalibrating the Genome-wide Association Null Hypothesis 2 Enables the Detection of Core Genes Underlying Complex 3 Traits 4 1,2 1,2 2-4 5 Wei Cheng , Sohini Ramachandran y, and Lorin Crawford y 6 1 Department of Ecology and Evolutionary Biology, Brown University, Providence, RI, 7 USA 8 2 Center for Computational Molecular Biology, Brown University, Providence, RI, USA 9 3 Department of Biostatistics, Brown University, Providence, RI, USA 10 4 Center for Statistical Sciences, Brown University, Providence, RI, USA 11 E-mail: [email protected]; lorin [email protected] y 12 Abstract 13 Traditional univariate genome-wide association studies generate false positives and negatives due to 14 difficulties distinguishing causal variants from \interactive" variants (i.e., variants correlated with causal 15 variants without directly influencing the trait). Recent efforts have been directed at identifying gene or 16 pathway associations, but these can be computationally costly and hampered by strict model assumptions. 17 Here, we present gene-", a new approach for identifying statistical associations between sets of variants 18 and quantitative traits. Our key innovation is a recalibration of the genome-wide null hypothesis to treat 19 small-yet-nonzero associations emitted by interactive variants as non-causal. gene-" efficiently identifies 20 core genes under a variety of simulated genetic architectures, achieving greater than a 90% true positive 21 rate at 1% false positive rate for polygenic traits. -

THE OLD and NEW FACE of CRANIOFACIAL RESEARCH: How Animal Models Inform Human Craniofacial Genetic and Clinical Data
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Dev Biol Manuscript Author . Author manuscript; Manuscript Author available in PMC 2017 July 15. Published in final edited form as: Dev Biol. 2016 July 15; 415(2): 171–187. doi:10.1016/j.ydbio.2016.01.017. THE OLD AND NEW FACE OF CRANIOFACIAL RESEARCH: How animal models inform human craniofacial genetic and clinical data Eric Van Otterloo, Trevor Williams, and Kristin B. Artinger Department of Craniofacial Biology, School of Dental Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA Kristin B. Artinger: [email protected] Abstract The craniofacial skeletal structures that comprise the human head develop from multiple tissues that converge to form the bones and cartilage of the face. Because of their complex development and morphogenesis, many human birth defects arise due to disruptions in these cellular populations. Thus, determining how these structures normally develop is vital if we are to gain a deeper understanding of craniofacial birth defects and devise treatment and prevention options. In this review, we will focus on how animal model systems have been used historically and in an ongoing context to enhance our understanding of human craniofacial development. We do this by first highlighting “animal to man” approaches: that is, how animal models are being utilized to understand fundamental mechanisms of craniofacial development. We discuss emerging technologies, including high throughput sequencing and genome editing, and new animal repository resources, and how their application can revolutionize the future of animal models in craniofacial research. Secondly, we highlight “man to animal” approaches, including the current use of animal models to test the function of candidate human disease variants. -

Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases
International Journal of Molecular Sciences Review Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases (DYRKs) and cdc2-Like Kinases (CLKs) in Human Disease, an Overview Mattias F. Lindberg and Laurent Meijer * Perha Pharmaceuticals, Perharidy Peninsula, 29680 Roscoff, France; [email protected] * Correspondence: [email protected] Abstract: Dual-specificity tyrosine phosphorylation-regulated kinases (DYRK1A, 1B, 2-4) and cdc2- like kinases (CLK1-4) belong to the CMGC group of serine/threonine kinases. These protein ki- nases are involved in multiple cellular functions, including intracellular signaling, mRNA splicing, chromatin transcription, DNA damage repair, cell survival, cell cycle control, differentiation, ho- mocysteine/methionine/folate regulation, body temperature regulation, endocytosis, neuronal development, synaptic plasticity, etc. Abnormal expression and/or activity of some of these kinases, DYRK1A in particular, is seen in many human nervous system diseases, such as cognitive deficits associated with Down syndrome, Alzheimer’s disease and related diseases, tauopathies, demen- tia, Pick’s disease, Parkinson’s disease and other neurodegenerative diseases, Phelan-McDermid syndrome, autism, and CDKL5 deficiency disorder. DYRKs and CLKs are also involved in dia- betes, abnormal folate/methionine metabolism, osteoarthritis, several solid cancers (glioblastoma, breast, and pancreatic cancers) and leukemias (acute lymphoblastic leukemia, acute megakaryoblas- Citation: Lindberg, M.F.; Meijer, L. tic leukemia), viral infections (influenza, HIV-1, HCMV, HCV, CMV, HPV), as well as infections Dual-Specificity, Tyrosine caused by unicellular parasites (Leishmania, Trypanosoma, Plasmodium). This variety of pathological Phosphorylation-Regulated Kinases implications calls for (1) a better understanding of the regulations and substrates of DYRKs and (DYRKs) and cdc2-Like Kinases CLKs and (2) the development of potent and selective inhibitors of these kinases and their evaluation (CLKs) in Human Disease, an as therapeutic drugs. -

AUTS2 Controls Neuronal Lineage Choice Through a Novel PRC1-Independent Complex And
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.29.450402; this version posted June 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. AUTS2 controls neuronal lineage choice through a novel PRC1-independent complex and BMP inhibition Zhuangzhuang Geng1, Qiang Wang1, Weili Miao2, Trevor Wolf3, Jessenia Chavez1, Emily Giddings3, Ryan Hobbs4, David J. DeGraff5,6, Yinsheng Wang2, James Stafford3, Zhonghua Gao1,6,7,# 1, Departments of Biochemistry and Molecular Biology, Penn State College of Medicine, Hershey, PA 17033, USA 2, Department of Chemistry, University of California at Riverside, Riverside, CA 92521, USA 3, Department of Neurological Sciences, Larner College of Medicine, University of Vermont, Burlington, VT 05405, USA 4, Department of Dermatology, Penn State College of Medicine, Hershey, PA 17033, USA 5, Department of Pathology and Laboratory Medicine, Penn State College of Medicine, Hershey, PA 17033, USA 6, Penn State Hershey Cancer Institute, Hershey, PA 17033, USA 7, The Stem Cell and Regenerative Biology Program, Penn State College of Medicine, Hershey, PA 17033, USA #, correspondence author, [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.06.29.450402; this version posted June 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary Despite a prominent risk factor for Neurodevelopmental disorders (NDD), it remains unclear how Autism Susceptibility Candidate 2 (AUTS2) controls the neurodevelopmental program. Our studies investigated the role of AUTS2 in neuronal differentiation and discovered that AUTS2, together with WDR68 and SKI, forms a novel protein complex (AWS) specifically in neuronal progenitors and promotes neuronal differentiation through inhibiting BMP signaling. -
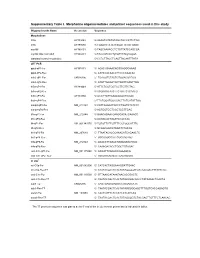
Supplementary Table I. Morpholino Oligonucleotides and Primer Sequences Used in This Study
Supplementary Table I. Morpholino oligonucleotides and primer sequences used in this study Oligonucleotide Name Accession Sequence Morpholinos tlr5a AY389449 5'-AAAGTGTATGTAGCTGCCATTCTGG tlr5b AY389450 5'-TGAATGTATATCCCATTCTGTGAGC myd88 AY388401 5'-TAGCAAAACCTCTGTTATCCAGCGA myd88 5bp mismatch AY388401 5'-TAcCAtAACCTgTGTTATCgAGgGA standard control morpholino 5'-CCTCTTACCTCAGTTACAATTTATA qRT-PCR ppial-qP1-Fw AY391451 5’- ACACTGAAACACGGAGGCAAAG ppial-qP2-Rev 5’- CATCCACAACCTTCCCGAACAC irak3-qP1-Fw CK026195 5’- TGAGGTCTACTGTGGACGATGG irak3-qP2-Rev 5’- ATGTTAGGATGCTGGTTGAGTTGG tlr5a-qP1-Fw AY389449 5’-ATTCTGGTGGTGCTTGTTGTAG tlr5a-qP2-Rev 5’-ACGAGGTAACTTCTGTTCTCAATG tlr5b-qP3-Fw AY389450 5’-GCGTTGTTGAAGAGGCTGGAC tlr5b-qP4-Rev 5’-TTCTGGATGGCCACTTCTCATATTGG mmp9-qP3-Fw NM_213123 5’-CATTAAAGATGCCCTGATGTATCCC mmp9-qP4-Rev 5’-AGTGGTGGTCCGTGGTTGAG il1b-qP1-Fw NM_212844 5’-GAACAGAATGAAGCACATCAAACC il1b-qP2-Rev 5’-ACGGCACTGAATCCACCAC il8-qP1-Fw XM_001342570 5’-TGTGTTATTGTTTTCCTGGCATTTC il8-qP2-Rev 5’-GCGACAGCGTGGATCTACAG ifn1-qP3-Fw NM_207640 5’- TTAATACACGCAAAGATGAGAACTC ifn1-qP4-Rev 5’- GCCAAGCCATTCGCAAGTAG tnfa-qP5-Fw NM_212829 5’- AGACCTTAGACTGGAGAGATGAC tnfa-qP6-Rev 5’- CAAAGACACCTGGCTGTAGAC cxcl-C1c-qP1-Fw NM_001115060 5’- GGCATTCACACCCAAAGCG cxcl-C1c-qP2_Rev 5’- GCGAGCACGATTCACGAGAG * In situ ccl-C5a-Fw NM_001082906 5’- CATCACTAGGAAAGGATTGAAC ccl-C5a-Rev-T7 5’- TAATACGACTCACTATAGGGGATGTCAAAGACTTTATTCAC cxcl-C1c-Fw NM_001115060 5’- GTTAAACATAAATAACACCGACTC cxcl-C1c-Rev-T7 5’- TAATACGACTCACTATAGGGACACCCTATAAAACTGAGTA irak3-Fw CK026195 5’- CAGTGAGAGAGGCATGAAACATC -

The Drosophila Wings Apart Gene Anchors a Novel, Evolutionarily Conserved Pathway of Neuromuscular Development
INVESTIGATION The Drosophila wings apart Gene Anchors a Novel, Evolutionarily Conserved Pathway of Neuromuscular Development Ginny R. Morriss, Carmelita T. Jaramillo, Crystal M. Mikolajczak, Sandy Duong, MaryAnn S. Jaramillo, and Richard M. Cripps1 Department of Biology, University of New Mexico, Albuquerque, New Mexico 87131 ABSTRACT wings apart (wap) is a recessive, semilethal gene located on the X chromosome in Drosophila melanogaster, which is required for normal wing-vein patterning. We show that the wap mutation also results in loss of the adult jump muscle. We use complementation mapping and gene-specific RNA interference to localize the wap locus to the proximal X chromosome. We identify the annotated gene CG14614 as the gene affected by the wap mutation, since one wap allele contains a non-sense mutation in CG14614, and a genomic fragment containing only CG14614 rescues the jump-muscle phenotypes of two wap mutant alleles. The wap gene lies centromere-proximal to touch-insensitive larva B and centromere-distal to CG14619, which is tentatively assigned as the gene affected in introverted mutants. In mutant wap animals, founder cell precursors for the jump muscle are specified early in development, but are later lost. Through tissue-specific knockdowns, we demonstrate that wap function is required in both the musculature and the nervous system for normal jump-muscle formation. wap/CG14614 is homologous to vertebrate wdr68, DDB1 and CUL4 associated factor 7, which also are expressed in neuromuscular tissues. Thus, our findings provide insight into mechanisms of neuromuscular development in higher animals and facilitate the understanding of neuromuscular diseases that may result from mis- expression of muscle-specific or neuron-specific genes. -

Green Tea Extracts Containing Epigallocatechin-3-Gallate
www.nature.com/scientificreports OPEN Green tea extracts containing epigallocatechin‑3‑gallate modulate facial development in Down syndrome John M. Starbuck1,2, Sergi Llambrich3, Rubèn Gonzàlez4, Julia Albaigès5,6,7, Anna Sarlé4, Jens Wouters3, Alejandro González8, Xavier Sevillano8, James Sharpe5,9,10, Rafael De La Torre11,12, Mara Dierssen5,6,7,13, Greetje Vande Velde3,13 & Neus Martínez‑Abadías4,5,10,13* Trisomy of human chromosome 21 (Down syndrome, DS) alters development of multiple organ systems, including the face and underlying skeleton. Besides causing stigmata, these facial dysmorphologies can impair vital functions such as hearing, breathing, mastication, and health. To investigate the therapeutic potential of green tea extracts containing epigallocatechin‑3‑gallate (GTE‑EGCG) for alleviating facial dysmorphologies associated with DS, we performed an experimental study with continued pre‑ and postnatal treatment with two doses of GTE‑EGCG supplementation in a mouse model of DS, and an observational study of children with DS whose parents administered EGCG as a green tea supplement. We evaluated the efect of high (100 mg/kg/day) or low doses (30 mg/kg/day) of GTE‑EGCG, administered from embryonic day 9 to post‑natal day 29, on the facial skeletal development in the Ts65Dn mouse model. In a cross‑sectional observational study, we assessed the facial shape in DS and evaluated the efects of self‑medication with green tea extracts in children from 0 to 18 years old. The main outcomes are 3D quantitative morphometric measures of the face, acquired either with micro‑computed tomography (animal study) or photogrammetry (human study). The lowest experimentally tested GTE‑EGCG dose improved the facial skeleton morphology in a mouse model of DS. -

Exon-Level Expression Profiling and Alternative Splicing in Autism Using Lymphoblastoid Cell Lines Zohreh Talebizadeha, Richard Aldenderfera and Xue Wen Chenb
Original article 1 A proof-of-concept study: exon-level expression profiling and alternative splicing in autism using lymphoblastoid cell lines Zohreh Talebizadeha, Richard Aldenderfera and Xue Wen Chenb Objective Autism is a complex, heterogeneous Conclusion The paucity of autism brain samples and neurobehavioral disorder with many causes and varying extensive phenotypic heterogeneity of autism demands degrees of severity. Some genetic implications related finding ways to also identify autism-related genomic events to autism may involve gene-regulatory processes such in accessible nonbrain resources, which may contribute as alternative splicing. Here, we assess the feasibility in biomarker identifications. This proof-of-concept study of profiling exon-level gene expression in autism using shows that the analysis of alternative splicing in the Affymetrix Human exon 1.0 ST array. lymphoblastoid cell line samples has a potential to reveal at least a subset of brain-related deregulation of splicing Methods We examined lymphoblastoid cell line-derived machinery that might be implicated in autism. Psychiatr RNAs from five patients with autism compared with five Genet 24:1–9 c 2014 Wolters Kluwer Health | Lippincott controls. Williams & Wilkins. Results Analysis of variance and Bonferroni Psychiatric Genetics 2014, 24:1–9 multiple test correction identified 57 genes exhibiting differential exon-level expression, suggesting Keywords: alternative splicing, autism, differential expression, exon array, lymphoblastoid cell lines potential changes in the resultant alternatively spliced transcripts in autism compared with controls. Genes aSection of Medical Genetics and Molecular Medicine, Children’s Mercy Hospital and University of Missouri–Kansas City School of Medicine, Kansas City, with differentially expressed exons included CYFIP1, Missouri and bDepartment of Electrical Engineering and Computer Science, a previously reported autism susceptibility gene.