Methanobrevibacter Sp. Abm4 S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methanobrevibacter Cuticularis Sp
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Oct. 1996, p. 3620–3631 Vol. 62, No. 10 0099-2240/96/$04.0010 Copyright q 1996, American Society for Microbiology Physiological Ecology of Methanobrevibacter cuticularis sp. nov. and Methanobrevibacter curvatus sp. nov., Isolated from the Hindgut of the Termite Reticulitermes flavipes JARED R. LEADBETTER AND JOHN A. BREZNAK* Department of Microbiology and Center for Microbial Ecology, Michigan State University, East Lansing, Michigan 48824-1101 Received 11 January 1996/Accepted 15 July 1996 Two morphologically distinct, H2- and CO2-utilizing methanogens were isolated from gut homogenates of the subterranean termite, Reticulitermes flavipes (Kollar) (Rhinotermitidae). Strain RFM-1 was a short straight rod (0.4 by 1.2 mm), whereas strain RFM-2 was a slightly curved rod (0.34 by 1.6 mm) that possessed polar fibers. Their morphology, gram-positive staining reaction, resistance to cell lysis by chemical agents, and narrow range of utilizable substrates were typical of species belonging to the family Methanobacteriaceae. Analysis of the nearly complete sequences of the small-subunit rRNA-encoding genes confirmed this affiliation and supported their recognition as new species of Methanobrevibacter: M. cuticularis (RFM-1) and M. curvatus (RFM-2). The per cell rates of methanogenesis by strains RFM-1 and RFM-2 in vitro, taken together with their in situ population densities (ca. 106 cells z gut21; equivalent to 109 cells z ml of gut fluid21), could fully account for the rate of methane emission by the live termites. UV epifluorescence and electron microscopy confirmed that RFM-1- and RFM-2-type cells were the dominant methanogens in R. -

Methanothermus Fervidus Type Strain (V24S)
UC Davis UC Davis Previously Published Works Title Complete genome sequence of Methanothermus fervidus type strain (V24S). Permalink https://escholarship.org/uc/item/9367m39j Journal Standards in genomic sciences, 3(3) ISSN 1944-3277 Authors Anderson, Iain Djao, Olivier Duplex Ngatchou Misra, Monica et al. Publication Date 2010-11-20 DOI 10.4056/sigs.1283367 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Standards in Genomic Sciences (2010) 3:315-324 DOI:10.4056/sigs.1283367 Complete genome sequence of Methanothermus fervidus type strain (V24ST) Iain Anderson1, Olivier Duplex Ngatchou Djao2, Monica Misra1,3, Olga Chertkov1,3, Matt Nolan1, Susan Lucas1, Alla Lapidus1, Tijana Glavina Del Rio1, Hope Tice1, Jan-Fang Cheng1, Roxanne Tapia1,3, Cliff Han1,3, Lynne Goodwin1,3, Sam Pitluck1, Konstantinos Liolios1, Natalia Ivanova1, Konstantinos Mavromatis1, Natalia Mikhailova1, Amrita Pati1, Evelyne Brambilla4, Amy Chen5, Krishna Palaniappan5, Miriam Land1,6, Loren Hauser1,6, Yun-Juan Chang1,6, Cynthia D. Jeffries1,6, Johannes Sikorski4, Stefan Spring4, Manfred Rohde2, Konrad Eichinger7, Harald Huber7, Reinhard Wirth7, Markus Göker4, John C. Detter1, Tanja Woyke1, James Bristow1, Jonathan A. Eisen1,8, Victor Markowitz5, Philip Hugenholtz1, Hans-Peter Klenk4, and Nikos C. Kyrpides1* 1 DOE Joint Genome Institute, Walnut Creek, California, USA 2 HZI – Helmholtz Centre for Infection Research, Braunschweig, Germany 3 Los Alamos National Laboratory, Bioscience Division, Los Alamos, New Mexico, USA 4 DSMZ - German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany 5 Biological Data Management and Technology Center, Lawrence Berkeley National Laboratory, Berkeley, California, USA 6 Oak Ridge National Laboratory, Oak Ridge, Tennessee, USA 7 University of Regensburg, Archaeenzentrum, Regensburg, Germany 8 University of California Davis Genome Center, Davis, California, USA *Corresponding author: Nikos C. -

Description of Methanobrevibacter Gottschalkii Sp. Nov
International Journal of Systematic and Evolutionary Microbiology (2002), 52, 819–822 DOI: 10.1099/ijs.0.02022-0 Description of Methanobrevibacter gottschalkii NOTE sp. nov., Methanobrevibacter thaueri sp. nov., Methanobrevibacter woesei sp. nov. and Methanobrevibacter wolinii sp. nov. Wadsworth Center, New Terry L. Miller and Chuzhao Lin York State Department of Health, Albany, NY 12201-0509, USA Author for correspondence: Terry L. Miller. Tel: j1 518 474 0015. Fax: j1 518 474 8590. e-mail: terry.miller!wadsworth.org Formal nomenclature is proposed for five methanogens, isolated from horse, pig, cow, goose and sheep faeces, that represent four novel species of the genus Methanobrevibacter. The four species, Methanobrevibacter gottschalkii sp. nov., Methanobrevibacter thaueri sp. nov., Methanobrevibacter woesei sp. nov. and Methanobrevibacter wolinii sp. nov., are distinguished from each other by a lack of genomic DNA reassociation and from previously described members of the genus on the basis of differences in the sequences of the 16S rRNA genes. Keywords: Methanobrevibacter, faecal methanogens Six of the seven species of Methanobrevibacter were (Conway de Macario et al., 1987; Ko$ nig, 1986; Miller isolated from gastrointestinal ecosystems (Miller, et al., 1986). 2001). Methanobrevibacter ruminantium, the type spe- The coccobacilli isolated from animals are distingui- cies, was isolated from the bovine rumen. Methano- shed from M. arboriphilicus, M. cuticularis, M. curva- brevibacter smithii is the predominant CO -reducing # tus and M. filiformis on the basis of differences in methanogen isolated from the human colon (Miller et morphology and physiology (Table 1). However, the al., 1986; Lin & Miller, 1998). The type strain of M. coccobacilli species of the genus Methanobrevibacter smithii was originally isolated from a municipal sewage and the animal isolates are not easily distinguishable digestor and was probably present in human faeces on the basis of biochemical and physiological charac- entering the treatment facility. -

Review Article: Inhibition of Methanogenic Archaea by Statins As a Targeted Management Strategy for Constipation and Related Disorders
Alimentary Pharmacology and Therapeutics Review article: inhibition of methanogenic archaea by statins as a targeted management strategy for constipation and related disorders K. Gottlieb*, V. Wacher*, J. Sliman* & M. Pimentel† *Synthetic Biologics, Inc., Rockville, SUMMARY MD, USA. † Gastroenterology, Cedars-Sinai Background Medical Center, Los Angeles, CA, USA. Observational studies show a strong association between delayed intestinal transit and the production of methane. Experimental data suggest a direct inhibitory activity of methane on the colonic and ileal smooth muscle and Correspondence to: a possible role for methane as a gasotransmitter. Archaea are the only con- Dr K. Gottlieb, Synthetic Biologics, fi Inc., 9605 Medical Center Drive, rmed biological sources of methane in nature and Methanobrevibacter Rockville, MD 20850, USA. smithii is the predominant methanogen in the human intestine. E-mail: [email protected] Aim To review the biosynthesis and composition of archaeal cell membranes, Publication data archaeal methanogenesis and the mechanism of action of statins in this context. Submitted 8 September 2015 First decision 29 September 2015 Methods Resubmitted 7 October 2015 Narrative review of the literature. Resubmitted 20 October 2015 Accepted 20 October 2015 Results EV Pub Online 11 November 2015 Statins can inhibit archaeal cell membrane biosynthesis without affecting This uncommissioned review article was bacterial numbers as demonstrated in livestock and humans. This opens subject to full peer-review. the possibility of a therapeutic intervention that targets a specific aetiologi- cal factor of constipation while protecting the intestinal microbiome. While it is generally believed that statins inhibit methane production via their effect on cell membrane biosynthesis, mediated by inhibition of the HMG- CoA reductase, there is accumulating evidence for an alternative or addi- tional mechanism of action where statins inhibit methanogenesis directly. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

Histone Variants in Archaea and the Evolution of Combinatorial Chromatin Complexity
Histone variants in archaea and the evolution of combinatorial chromatin complexity Kathryn M. Stevensa,b, Jacob B. Swadlinga,b, Antoine Hochera,b, Corinna Bangc,d, Simonetta Gribaldoe, Ruth A. Schmitzc, and Tobias Warneckea,b,1 aMolecular Systems Group, Quantitative Biology Section, Medical Research Council London Institute of Medical Sciences, London W12 0NN, United Kingdom; bInstitute of Clinical Sciences, Faculty of Medicine, Imperial College London, London W12 0NN, United Kingdom; cInstitute for General Microbiology, University of Kiel, 24118 Kiel, Germany; dInstitute of Clinical Molecular Biology, University of Kiel, 24105 Kiel, Germany; and eDepartment of Microbiology, Unit “Evolutionary Biology of the Microbial Cell,” Institut Pasteur, 75015 Paris, France Edited by W. Ford Doolittle, Dalhousie University, Halifax, NS, Canada, and approved October 28, 2020 (received for review April 14, 2020) Nucleosomes in eukaryotes act as platforms for the dynamic inte- additional histone dimers can be taggedontothistetramertoyield gration of epigenetic information. Posttranslational modifications oligomers of increasing length that wrap correspondingly more DNA are reversibly added or removed and core histones exchanged for (3, 6–9). Almost all archaeal histones lack tails and PTMs have yet to paralogous variants, in concert with changing demands on tran- be reported. Many archaea do, however, encode multiple histone scription and genome accessibility. Histones are also common in paralogs (8, 10) that can flexibly homo- and heterodimerize in -

Supporting Information
Supporting Information Lozupone et al. 10.1073/pnas.0807339105 SI Methods nococcus, and Eubacterium grouped with members of other Determining the Environmental Distribution of Sequenced Genomes. named genera with high bootstrap support (Fig. 1A). One To obtain information on the lifestyle of the isolate and its reported member of the Bacteroidetes (Bacteroides capillosus) source, we looked at descriptive information from NCBI grouped firmly within the Firmicutes. This taxonomic error was (www.ncbi.nlm.nih.gov/genomes/lproks.cgi) and other related not surprising because gut isolates have often been classified as publications. We also determined which 16S rRNA-based envi- Bacteroides based on an obligate anaerobe, Gram-negative, ronmental surveys of microbial assemblages deposited near- nonsporulating phenotype alone (6, 7). A more recent 16S identical sequences in GenBank. We first downloaded the gbenv rRNA-based analysis of the genus Clostridium defined phylo- files from the NCBI ftp site on December 31, 2007, and used genetically related clusters (4, 5), and these designations were them to create a BLAST database. These files contain GenBank supported in our phylogenetic analysis of the Clostridium species in the HGMI pipeline. We thus designated these Clostridium records for the ENV database, a component of the nonredun- species, along with the species from other named genera that dant nucleotide database (nt) where 16S rRNA environmental cluster with them in bootstrap supported nodes, as being within survey data are deposited. GenBank records for hits with Ͼ98% these clusters. sequence identity over 400 bp to the 16S rRNA sequence of each of the 67 genomes were parsed to get a list of study titles Annotation of GTs and GHs. -

Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China
Characteristics and Metabolic Patterns of Soil Methanogenic Archaea Communities in the High Latitude Natural Wetlands of China Di Wu Northeast Forestry University Caihong Zhao Northeast Forestry University Hui Bai Forestry Science Research Institute of Heilongjiang Province Fujuan Feng Northeast Forestry University Xin Sui Heilongjiang University Guangyu Sun ( [email protected] ) Northeast Forestry University Research article Keywords: Wetlands, Methanogens, Community diversity, Indicator species, Methanogenic metabolic patterns Posted Date: August 12th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-54821/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Background: Soil methanogenic microorganisms are one of the primary methane-producing microbes in wetlands. However, we still poorly understand the community characteristic and metabolic patterns of these microorganisms according to vegetation type and seasonal changes. Therefore, to better elucidate the effects of the vegetation type and seasonal factors on the methanogenic community structure and metabolic patterns, we detected the characteristics of the soil methanogenic mcrA gene from three types of natural wetlands in different seasons in the Xiaoxing'an Mountain region, China. Result: The results indicated that the distribution of Methanobacteriaceae (hydrogenotrophic methanogens) was higher in winter, while Methanosarcinaceae and Methanosaetaceae accounted for a higher proportion in summer. Hydrogenotrophic methanogenesis was the dominant trophic pattern in each wetland. The results of principal coordinate analysis and cluster analysis showed that the vegetation type considerably inuenced the methanogenic community composition. The methanogenic community structure in the Betula platyphylla – Larix gmelinii wetland was relatively different from the structure of the other two wetland types. -

Development of the Equine Hindgut Microbiome in Semi-Feral and Domestic Conventionally-Managed Foals Meredith K
Tavenner et al. Animal Microbiome (2020) 2:43 Animal Microbiome https://doi.org/10.1186/s42523-020-00060-6 RESEARCH ARTICLE Open Access Development of the equine hindgut microbiome in semi-feral and domestic conventionally-managed foals Meredith K. Tavenner1, Sue M. McDonnell2 and Amy S. Biddle1* Abstract Background: Early development of the gut microbiome is an essential part of neonate health in animals. It is unclear whether the acquisition of gut microbes is different between domesticated animals and their wild counterparts. In this study, fecal samples from ten domestic conventionally managed (DCM) Standardbred and ten semi-feral managed (SFM) Shetland-type pony foals and dams were compared using 16S rRNA sequencing to identify differences in the development of the foal hindgut microbiome related to time and management. Results: Gut microbiome diversity of dams was lower than foals overall and within groups, and foals from both groups at Week 1 had less diverse gut microbiomes than subsequent weeks. The core microbiomes of SFM dams and foals had more taxa overall, and greater numbers of taxa within species groups when compared to DCM dams and foals. The gut microbiomes of SFM foals demonstrated enhanced diversity of key groups: Verrucomicrobia (RFP12), Ruminococcaceae, Fusobacterium spp., and Bacteroides spp., based on age and management. Lactic acid bacteria Lactobacillus spp. and other Lactobacillaceae genera were enriched only in DCM foals, specifically during their second and third week of life. Predicted microbiome functions estimated computationally suggested that SFM foals had higher mean sequence counts for taxa contributing to the digestion of lipids, simple and complex carbohydrates, and protein. -

Susceptibility of Archaea to Antimicrobial Agents: Applications to Clinical Microbiology
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REVIEW 10.1111/j.1469-0691.2012.03913.x Susceptibility of archaea to antimicrobial agents: applications to clinical microbiology S. Khelaifia and M. Drancourt Unite´ de Recherche sur les Maladies Infectieuses et Tropicales Emergentes, UMR CNRS 6236 IRD 3R198, Me´diterrane´e Infection, Faculte´ de Me´decine, Aix-marseille-Universite´, Marseille, France Abstract We herein review the state of knowledge regarding the in vitro and in vivo susceptibility of archaea to antimicrobial agents, including some new molecules. Indeed, some archaea colonizing the human microbiota have been implicated in diseases such as periodontopathy. Archaea are characterized by their broad-spectrum resistance to antimicrobial agents. In particular, their cell wall lacks peptidoglycan, making them resistant to antimicrobial agents interfering with peptidoglycan biosynthesis. Archaea are, however, susceptible to the pro- tein synthesis inhibitor fusidic acid and imidazole derivatives. Also, squalamine, an antimicrobial agent acting on the cell wall, proved effective against human methanogenic archaea. In vitro susceptibility data could be used to design protocols for the decontamination of complex microbiota and the selective isolation of archaea in anaerobic culture. Keywords: Antimicrobial agent, archaea, methanogenic archaea, microbiota, susceptibility testing Article published online: 23 May 2012 Clin Microbiol Infect 2012; 18: 841–848 Corresponding author: M. Drancourt, Unite´ des Rickettsies, Fa- culte´ de Me´decine, 27, Boulevard Jean Moulin-Cedex 5, France E-mail: [email protected] Methanogenic archaea (herein referred to as methano- Introduction gens) are the sole organisms producing methane from H2 +CO2 [6]. -

Evaluation of Methanobrevibacter Smithii As a Human-Specific Marker
The University of Southern Mississippi The Aquila Digital Community Presentations Microbial Source Tracking 2005 Evaluation of Methanobrevibacter smithii as a Human-Specific aM rker of Fecal Pollution Jennifer A. Ufnar University of Southern Mississippi Shiao Y. Wang University of Southern Mississippi R.D. Ellender University of Southern Mississippi Follow this and additional works at: https://aquila.usm.edu/mst_presentations Recommended Citation Ufnar, Jennifer A.; Wang, Shiao Y.; and Ellender, R.D., "Evaluation of Methanobrevibacter smithii as a Human-Specific aM rker of Fecal Pollution" (2005). Presentations. 5. https://aquila.usm.edu/mst_presentations/5 This Poster is brought to you for free and open access by the Microbial Source Tracking at The Aquila Digital Community. It has been accepted for inclusion in Presentations by an authorized administrator of The Aquila Digital Community. For more information, please contact [email protected]. Evaluation of Methanobrevibacter smithii as a Human-Specific Q-311 For More Information Contact: Marker of Fecal Pollution Dr. R.D. Ellender University of Southern Mississippi Department of Biological Sciences Jennifer A. Ufnar, Shiao Y. Wang, and R.D. Ellender 118 College Drive #5018 Hattiesburg, MS 39406-0001 University of Southern Mississippi, Hattiesburg, MS 601-266-4720 or 601-266-4752(lab) Results Abstract Primers specific for the Methanobrevibacter genus (MET) amplified a product of 282bp. The Mnif primers amplified a 222bp product. The MET primers were Microbial source tracking has historically focused on the origin of traditional enteric indicators including coliforms, enterococci, or Escherichia coli. Recently, positive for all Methanobrevibacter species, and did not amplify a product in any other bacterial or methanogen genus. -
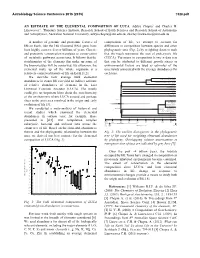
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3].