Specific NEMO Mutations Impair CD40-Mediated C-Rel Activation and B Cell Terminal Differentiation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cathepsins and Their Involvement in Immune Responses
Review article: Medical intelligence | Published 20 July 2010, doi:10.4414/smw.2010.13042 Cite this as: Swiss Med Wkly. 2010;140:w13042 Cathepsins and their involvement in immune responses Sébastien Conus, Hans-Uwe Simon Institute of Pharmacology, University of Bern, Bern, Switzerland Correspondence to: cleaving proteases (= caspases) which cleave a wide range Sébastien Conus Ph.D. of cellular substrates [5]. Institute of Pharmacology Besides caspases, cathepsins have recently been shown University of Bern to be associated with cell death regulation [6–12] and vari- Friedbühlstrasse 49 3010 Bern ous other physiological and pathological processes, such as Switzerland maturation of the MHC class II complex, bone remodel- [email protected] ling, keratinocyte differentiation, tumour progression and metastasis, rheumatoid arthritis and osteoarthritis, as well Summary as atherosclerosis [13, 14] (table 1). Thus, cathepsins ap- pear to play a significant role in immune responses. In this The immune system is composed of an enormous variety of review we discuss recent advances addressing the role of cells and molecules that generate a collective and coordin- lysosomal proteases in the diverse aspects of the immune ated response on exposure to foreign antigens, called the response, and also the involvement of cathepsins in the immune response. Within the immune response, endo-lyso- pathogenesis of diseases in which these proteases seem not somal proteases play a key role. In this review we cover to be properly under control. specific roles of cathepsins in innate and adaptive immu- nity, as well as their implication in the pathogenesis of sev- The cathepsin family eral diseases. Lysosomes are membrane-bound organelles which repres- Key words: adaptive and innate immunity; apoptosis; ent the main degradative compartment in eukaryotic cells. -

A Cysteine Protease Inhibitor Blocks SARS-Cov-2 Infection of Human and Monkey Cells
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.23.347534; this version posted October 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. A cysteine protease inhibitor blocks SARS-CoV-2 infection of human and monkey cells Drake M. Mellott,1 Chien-Te Tseng,3 Aleksandra Drelich,3 Pavla Fajtová,4,5 Bala C. Chenna,1 Demetrios H. Kostomiris1, Jason Hsu,3 Jiyun Zhu,1 Zane W. Taylor,2,9 Vivian Tat,3 Ardala Katzfuss,1 Linfeng Li,1 Miriam A. Giardini,4 Danielle Skinner,4 Ken Hirata,4 Sungjun Beck4, Aaron F. Carlin,8 Alex E. Clark4, Laura Beretta4, Daniel Maneval6, Felix Frueh,6 Brett L. Hurst,7 Hong Wang,7 Klaudia I. Kocurek,2 Frank M. Raushel,2 Anthony J. O’Donoghue,4 Jair Lage de Siqueira-Neto,4 Thomas D. Meek1.*, and James H. McKerrow#4,* Departments of Biochemistry and Biophysics1 and Chemistry,2 Texas A&M University, 301 Old Main Drive, College Station, Texas 77843, 3Department of Microbiology and Immunology, University of Texas, Medical Branch, 3000 University Boulevard, Galveston, Texas, 77755-1001, 4Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA, 5Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, 16610 Prague, Czech Republic, 6Selva Therapeutics, and 7Institute for Antiviral Research, Department of Animal, Dairy, and Veterinary Sciences, 5600 Old Main Hill, Utah State University, Logan, Utah, 84322, 8Department of Medicine, Division of Infectious Diseases and Global Public Health, University of California, San Diego, La Jolla, CA 92037, USA.9Current address: Biological Sciences Division, Pacific Northwest National Laboratory, 902 Battelle Blvd, Richland, WA 99353. -

Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients with Stable Coronary Heart Disease
Supplementary Online Content Ganz P, Heidecker B, Hveem K, et al. Development and validation of a protein-based risk score for cardiovascular outcomes among patients with stable coronary heart disease. JAMA. doi: 10.1001/jama.2016.5951 eTable 1. List of 1130 Proteins Measured by Somalogic’s Modified Aptamer-Based Proteomic Assay eTable 2. Coefficients for Weibull Recalibration Model Applied to 9-Protein Model eFigure 1. Median Protein Levels in Derivation and Validation Cohort eTable 3. Coefficients for the Recalibration Model Applied to Refit Framingham eFigure 2. Calibration Plots for the Refit Framingham Model eTable 4. List of 200 Proteins Associated With the Risk of MI, Stroke, Heart Failure, and Death eFigure 3. Hazard Ratios of Lasso Selected Proteins for Primary End Point of MI, Stroke, Heart Failure, and Death eFigure 4. 9-Protein Prognostic Model Hazard Ratios Adjusted for Framingham Variables eFigure 5. 9-Protein Risk Scores by Event Type This supplementary material has been provided by the authors to give readers additional information about their work. Downloaded From: https://jamanetwork.com/ on 10/02/2021 Supplemental Material Table of Contents 1 Study Design and Data Processing ......................................................................................................... 3 2 Table of 1130 Proteins Measured .......................................................................................................... 4 3 Variable Selection and Statistical Modeling ........................................................................................ -

Does the Use of Protease Inhibitors Further Improve in Vivo Distribution?
Targeting of the Cholecystokinin-2 Receptor with the Minigastrin Analog 177Lu-DOTA-PP-F11N: Does the Use of Protease Inhibitors Further Improve In Vivo Distribution? Alexander W. Sauter*1,2, Rosalba Mansi*3, Ulrich Hassiepen4, Lionel Muller4, Tania Panigada4, Stefan Wiehr2, Anna-Maria Wild2, Susanne Geistlich5, Martin B´eh´e5, Christof Rottenburger1, Damian Wild1, and Melpomeni Fani3 1Division of Nuclear Medicine, University Hospital Basel, Basel, Switzerland; 2Werner Siemens Imaging Center, Department of Preclinical Imaging and Radiopharmacy, Eberhard Karls University, Tuebingen, Germany; 3Division of Radiopharmaceutical Chemistry, University Hospital Basel, Basel, Switzerland; 4Novartis Pharma AG, Institutes for Biomedical Research, Novartis Campus, Basel, Switzerland; and 5Center for Radiopharmaceutical Sciences, Paul Scherrer Institute, Villigen, Switzerland performance of 177Lu-DOTA-MG11 in the presence of inhibitors. The Patients with metastatic medullary thyroid cancer (MTC) have limited human application of single compounds without unessential additives systemic treatment options. The use of radiolabeled gastrin analogs is preferable. Preliminary clinical data spotlight the stomach as a po- targeting the cholecystokinin-2 receptor (CCK2R) is an attractive tential dose-limiting organ besides the kidneys. approach. However, their therapeutic efficacy is presumably decreased Key Words: medullary thyroid cancer; cholecystokinin-2 receptor; by their enzymatic degradation in vivo. We aimed to investigate whether gastrin; peptide receptor radionuclide therapy; 177Lu-DOTA-PP-F11N 177 177 the chemically stabilized analog Lu-DOTA-PP-F11N ( Lu-DOTA- J Nucl Med 2019; 60:393–399 (DGlu)6-Ala-Tyr-Gly-Trp-Nle-Asp-Phe-NH2) performs better than refer- DOI: 10.2967/jnumed.118.207845 ence analogs with varying in vivo stability, namely 177Lu-DOTA-MG11 177 177 ( Lu-DOTA-DGlu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2)and Lu-DOTA- 177 PP-F11 ( Lu-DOTA-(DGlu)6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2), and whether the use of protease inhibitors further improves CCKR2 targeting. -

Death Penalty for Keratinocytes: Apoptosis Versus Cornification
Cell Death and Differentiation (2005) 12, 1497–1508 & 2005 Nature Publishing Group All rights reserved 1350-9047/05 $30.00 www.nature.com/cdd Review Death penalty for keratinocytes: apoptosis versus cornification S Lippens1,2, G Denecker1, P Ovaere1, P Vandenabeele*,1 and apoptosis, necrosis or autophagy ultimately result in the W Declercq*,1 elimination of particular cells from a tissue. However, in specialized forms of differentiation, dead cell corpses are not 1 Molecular Signaling and Cell Death Unit, Department for Molecular Biomedical removed but maintained to fulfil a specific function. These Research, VIB (Flanders Interuniversity Institute for Biotechnology) and Ghent developmental cell death programs result in the production of University, Technologiepark 927, B-9052 Zwijnaarde, Belgium 2 differentiated ‘storage’ cells containing large amounts of Current address: Institute de Biochimie, Universite´ de Lausanne, Chemin des specific proteins or other substances. Examples of such Boveresses 155, CH-1066 Epalinges, Switzerland * Corresponding authors: W Declercq and P Vandenabeele, Molecular Signaling differentiation programs occur in the stalk of the slime mold and Cell Death Unit, Department of Molecular Biomedical Research, Dictyostelium, during xylogenesis in plants, erythrocyte Technologiepark 927, B-9052 Zwijnaarde, Belgium. differentiation, lens fiber formation and cornification of Tel: þ 32 9 33137 60 Fax: þ 32 9 3313609; keratinocytes in the skin. E-mails: [email protected], Both apoptosis and keratinocyte cornification share some [email protected] similarities at the cellular and molecular level, such as loss of an intact nucleus and other organelles, cytoskeleton and cell Received 17.6.04; revised 23.3.05; accepted 07.4.05 Edited by G Melino shape changes, involvement of proteolytic events and mitochondrial changes. -

Fibroblasts from the Human Skin Dermo-Hypodermal Junction Are
cells Article Fibroblasts from the Human Skin Dermo-Hypodermal Junction are Distinct from Dermal Papillary and Reticular Fibroblasts and from Mesenchymal Stem Cells and Exhibit a Specific Molecular Profile Related to Extracellular Matrix Organization and Modeling Valérie Haydont 1,*, Véronique Neiveyans 1, Philippe Perez 1, Élodie Busson 2, 2 1, 3,4,5,6, , Jean-Jacques Lataillade , Daniel Asselineau y and Nicolas O. Fortunel y * 1 Advanced Research, L’Oréal Research and Innovation, 93600 Aulnay-sous-Bois, France; [email protected] (V.N.); [email protected] (P.P.); [email protected] (D.A.) 2 Department of Medical and Surgical Assistance to the Armed Forces, French Forces Biomedical Research Institute (IRBA), 91223 CEDEX Brétigny sur Orge, France; [email protected] (É.B.); [email protected] (J.-J.L.) 3 Laboratoire de Génomique et Radiobiologie de la Kératinopoïèse, Institut de Biologie François Jacob, CEA/DRF/IRCM, 91000 Evry, France 4 INSERM U967, 92260 Fontenay-aux-Roses, France 5 Université Paris-Diderot, 75013 Paris 7, France 6 Université Paris-Saclay, 78140 Paris 11, France * Correspondence: [email protected] (V.H.); [email protected] (N.O.F.); Tel.: +33-1-48-68-96-00 (V.H.); +33-1-60-87-34-92 or +33-1-60-87-34-98 (N.O.F.) These authors contributed equally to the work. y Received: 15 December 2019; Accepted: 24 January 2020; Published: 5 February 2020 Abstract: Human skin dermis contains fibroblast subpopulations in which characterization is crucial due to their roles in extracellular matrix (ECM) biology. -

Cathepsin C Independent Component That Requires
The IL-1−Dependent Sterile Inflammatory Response Has a Substantial Caspase-1− Independent Component That Requires Cathepsin C This information is current as of September 23, 2021. Hajime Kono, Gregory M. Orlowski, Zubin Patel and Kenneth L. Rock J Immunol published online 22 August 2012 http://www.jimmunol.org/content/early/2012/08/22/jimmun ol.1200136 Downloaded from Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 23, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published August 22, 2012, doi:10.4049/jimmunol.1200136 The Journal of Immunology The IL-1–Dependent Sterile Inflammatory Response Has a Substantial Caspase-1–Independent Component That Requires Cathepsin C Hajime Kono,* Gregory M. Orlowski,† Zubin Patel,† and Kenneth L. Rock† The sterile inflammatory response to cell death and irritant crystals is medically important because it causes disease. Although these stimuli are structurally distinct, they cause inflammation through a common pathway that requires the cytokine IL-1. -

Cathepsin C Independent Component That Requires
The IL-1−Dependent Sterile Inflammatory Response Has a Substantial Caspase-1− Independent Component That Requires Cathepsin C This information is current as of September 27, 2021. Hajime Kono, Gregory M. Orlowski, Zubin Patel and Kenneth L. Rock J Immunol 2012; 189:3734-3740; Prepublished online 22 August 2012; doi: 10.4049/jimmunol.1200136 Downloaded from http://www.jimmunol.org/content/189/7/3734 References This article cites 39 articles, 15 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/189/7/3734.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 27, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology The IL-1–Dependent Sterile Inflammatory Response Has a Substantial Caspase-1–Independent Component That Requires Cathepsin C Hajime Kono,* Gregory M. Orlowski,† Zubin Patel,† and Kenneth L. Rock† The sterile inflammatory response to cell death and irritant crystals is medically important because it causes disease. -

A Genomic Analysis of Rat Proteases and Protease Inhibitors
A genomic analysis of rat proteases and protease inhibitors Xose S. Puente and Carlos López-Otín Departamento de Bioquímica y Biología Molecular, Facultad de Medicina, Instituto Universitario de Oncología, Universidad de Oviedo, 33006-Oviedo, Spain Send correspondence to: Carlos López-Otín Departamento de Bioquímica y Biología Molecular Facultad de Medicina, Universidad de Oviedo 33006 Oviedo-SPAIN Tel. 34-985-104201; Fax: 34-985-103564 E-mail: [email protected] Proteases perform fundamental roles in multiple biological processes and are associated with a growing number of pathological conditions that involve abnormal or deficient functions of these enzymes. The availability of the rat genome sequence has opened the possibility to perform a global analysis of the complete protease repertoire or degradome of this model organism. The rat degradome consists of at least 626 proteases and homologs, which are distributed into five catalytic classes: 24 aspartic, 160 cysteine, 192 metallo, 221 serine, and 29 threonine proteases. Overall, this distribution is similar to that of the mouse degradome, but significatively more complex than that corresponding to the human degradome composed of 561 proteases and homologs. This increased complexity of the rat protease complement mainly derives from the expansion of several gene families including placental cathepsins, testases, kallikreins and hematopoietic serine proteases, involved in reproductive or immunological functions. These protease families have also evolved differently in the rat and mouse genomes and may contribute to explain some functional differences between these two closely related species. Likewise, genomic analysis of rat protease inhibitors has shown some differences with the mouse protease inhibitor complement and the marked expansion of families of cysteine and serine protease inhibitors in rat and mouse with respect to human. -
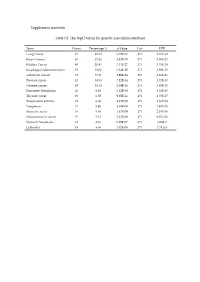
Supplement Materials Table S1. the Top15 Terms for Genetic Association
Supplement materials Table S1. The top15 terms for genetic association database. Term Count Percentage % p-Value List FDR Lung Cancer 73 25.34 6.70E-36 271 1.16E-32 Breast Cancer 67 23.26 2.92E-30 271 5.06E-27 Bladder Cancer 60 20.83 3.33E-27 271 5.78E-24 Esophageal adenocarcinoma 52 18.06 6.24E-29 271 1.08E-25 Colorectal Cancer 51 17.71 4.99E-24 271 8.66E-21 Prostate cancer 42 14.58 7.12E-14 271 1.23E-10 Ovarian cancer 39 13.54 2.04E-16 271 3.89E-13 Pancreatic Neoplasms 20 6.94 1.45E-08 271 2.52E-05 Thyroid cancer 19 6.59 9.98E-11 271 1.73E-07 Rheumatoid arthritis 19 6.60 8.82E-08 271 1.53E-04 Neoplasms 17 5.90 4.58E-08 271 7.94E-05 Stomach cancer 16 5.56 1.17E-09 271 2.03E-06 Head and neck cancer 15 5.21 5.23E-09 271 9.07E-06 Stomach Neoplasms 14 4.86 6.20E-07 271 1.08E-3 Leukemia 14 4.86 1.02E-06 271 1.78 E-3 Table S2. The results of the reverse docking for AutoDock Vina. PDB Protein Gene_Name Uniprot_ID Score 4TVJ Poly [ADP-ribose] polymerase 2 PARP2 Q9UGN5 -10.80 2DQ7 Proto-oncogene tyrosine-protein kinase Fyn FYN P06241 -10.00 4KIK Inhibitor of nuclear factor kappa-B kinase IKBKB O14920 -10.00 subunit beta 3EQR Activated CDC42 kinase 1 ACK1/TNK2 Q07912 -9.90 3MTF Activin receptor type-1 ACVR1 Q04771 -9.90 4GV0 Poly [ADP-ribose] polymerase 3 PARP3 Q9Y6F1 -9.90 1M6I Programmed cell death protein 8 AIF O95831 -9.80 1UA2 Cell division protein kinase 7 CDK7 P50613 -9.80 1Z6T Apoptotic protease activating factor 1 APAF1 O14727 -9.80 1ZXM DNA topoisomerase II TOPII P11388 -9.80 2WOU Serine/Threonine-protein kinase NEK7 NEK7 Q8TDX7 -9.60 -

Therapeutic Potential of Inhibition of the Neuroinflammation Induced Cathepsin X: in Vivo Evidence
bioRxiv preprint doi: https://doi.org/10.1101/513671; this version posted January 7, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Therapeutic potential of inhibition of the neuroinflammation induced cathepsin X: in vivo evidence Anja Pišlar1†*, Larisa Tratnjek2,3†, Gordana Glavan4, Nace Zidar5, Marko Živin2, Janko Kos1,6 1 Department of Pharmaceutical Biology, Faculty of Pharmacy, University of Ljubljana, Aškerčeva 7, 1000 Ljubljana, Slovenia 2 Institute of Pathophysiology, Medical faculty, University of Ljubljana, Zaloška 4, 1000 Ljubljana, Slovenia 3 Institute of Cell Biology, Medical faculty, University of Ljubljana, Vrazov trg 2, 1000 Ljubljana, Slovenia 4 Department of Biology, Biotechnical faculty, University of Ljubljana, Večna pot 11, 1000 Ljubljana, Slovenia 5 Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Ljubljana, Aškerčeva 7, 1000 Ljubljana, Slovenia 6 Department of Biotechnology, Jožef Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia Running title: Cathepsin X upregulation in the LPS rat model † These authors contributed equally. * Correspondence to: Anja Pišlar, Faculty of Pharmacy, University of Ljubljana, Aškerčeva 7, 1000 Ljubljana, Slovenia, Tel: +386 1 4769526, Fax: +386 1 4258031, E-mail: [email protected]. 1 bioRxiv preprint doi: https://doi.org/10.1101/513671; this version posted January 7, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Parkinson’s disease (PD) is a progressive neurodegenerative disorder with unknown cause, but it has been postulated that chronic neuroinflammation may play a role in its pathogenesis. -

Supplemental Material
Supplemental Figure 1. Heat map comparing expression of individual genes and significant modules. Expression values were normalized by gene and mean normalized expression values for each experimental animal (Control or SIVE) were calculated for the set of filtered significant modules. Compared with individual gene expression, average module expression yields a more homogeneous, though less intense, differentiation between uninfected and SIVE animals. Figure was generated by TMeV software [http://www.tm4.org/mev.html] . Supplemental Figure 2. Multiple chemokine receptors are expressed on differentiated SK-N-MC cells. Cells were differentiated for 4 days with retinoic acid prior to FACS analysis for chemokine receptors CCR2 (ligands: CCL2, CCL8), CCR3 (CCL8), CXCR1 (IL8), CXCR2 (IL8), CXCR3 (CXCL10), and high- and low-affinity NGF receptors. Table S1. Summary of rhesus monkeys - treatment history CONTROL Monkey Age (years) Sex anti-CD8 treatment Days infected Brain Viral Load 298 5 Male No NA NA 331 6 Male No NA NA 332 5 Male No NA NA 349 3 Male No NA NA 357 4 Male No NA NA 359 8 Male No NA NA 382 5 Male No NA NA 392 4 Male Yes NA NA 393 3 Male Yes NA NA SIVE 289 6 Male Yes 72 5.41 321 6 Male Yes 108 5.77 323 6 Male Yes 93 6.68 328 6 Male Yes 79 6.20 383 5 Male No 132 6.06 417 2 Male No 56 5.89 418 5 Male No 82 6.76 427 4 Male No 104 6.63 494 5 Male No 105 5.96 Monkeys used in this study.