ABSTRACT Gamma Hydroxybutyrate (GHB): Mechanisms of Central
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
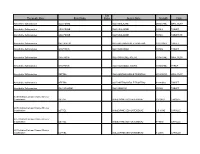
Therapeutic Class Brand Name P a Status Generic
P A Therapeutic Class Brand Name Status Generic Name Strength Form Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 250MG/5ML ORAL SUSP Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 500MG TABLET Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 500MG TABLET DR Absorbable Sulfonamides BACTRIM DS SULFAMETHOXAZOLE/TRIMETHO 800-160MG TABLET Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE 500MG TABLET Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE ACETYL 500MG/5ML ORAL SUSP Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE ACETYL 500MG/5ML SYRUP Absorbable Sulfonamides SEPTRA SULFAMETHOXAZOLE/TRIMETHO 200-40MG/5 ORAL SUSP Absorbable Sulfonamides SEPTRA SULFAMETHOXAZOLE/TRIMETHO 400-80MG TABLET Absorbable Sulfonamides SULFADIAZINE SULFADIAZINE 500MG TABLET ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 10-20MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 2.5-10MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-10MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-20MG CAPSULE P A Therapeutic Class Brand Name Status Generic Name Strength Form ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-40MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 10-40MG CAPSULE Acne Agents, Systemic ACCUTANE ISOTRETINOIN 10MG CAPSULE Acne Agents, Systemic ACCUTANE ISOTRETINOIN 20MG CAPSULE Acne Agents, Systemic ACCUTANE -

Summary: Traditionally, Bioanalytical Laboratories Do Not Report Actual
Summary: Traditionally, bioanalytical laboratories do not report actual concentrations for samples with results below the limit of quantification (BLQ) in pharmacokinetic studies. BLQ values are outside the method calibration range established during validation and no data are available to support the reliability of these values. However, ignoring BLQ data can contribute to bias and imprecision in model-based pharmacokinetic analyses. From this perspective, routine use of BLQ data would be advantageous. We would like to initiate an interdisciplinary debate on this important topic by summarising the current concepts and use of BLQ data by regulators, pharmacometricians and bioanalysts. Through introducing the limit of detection and evaluating its variability BLQ data could be released and utilized appropriately for pharmacokinetic research. Keywords: Lower limit of quantification (LLOQ) Below limit of quantification (BLQ) result Limit of detection (LoD) Pharmacokinetic (PK) Pharmacodynamics (PD) Introduction Studying the effects of drugs remains central to both medical research and clinical practice. Two key branches of pharmacological analysis are (i) pharmacokinetics (PK), including drug absorption, distribution, metabolism and elimination, and (ii) pharmacodynamics (PD), exploring the effects of drugs on the living organism, including efficacy and toxicity. In PK studies the samples are collected in an effort to map the drug concentration over time in the patient. For samples collected many hours post-dose drug concentrations may be low, yet can still provide valuable information on pharmacokinetic parameters such as clearance [1,2]. Similarly, in the case of biomarker PD studies, concentrations that are too low to quantify with a particular bioanalytical method may still provide useful information. Bioanalytical laboratories define the lowest concentration that can be quantified accurately by a method as the lower limit of quantification (LLOQ). -

(12) United States Patent (10) Patent No.: US 8,822,539 B2 Jensen (45) Date of Patent: Sep
USOO8822.539B2 (12) United States Patent (10) Patent No.: US 8,822,539 B2 Jensen (45) Date of Patent: Sep. 2, 2014 (54) COMBINATION THERAPIES: INHIBITORS Bialer et al., “Progress report on new antiepileptic drugs: a Summary OF GABA TRANSAMINASE AND NKCC1 of the Seventh Eilat Conference (EILATVII).” Epilepsy Res., 61:1- 48, 2004 (Abstract only). (75) Inventor: Frances E. Jensen, Boston, MA (US) Clift and Silverman, “Synthesis and Evaluation of Novel Aromatic Substrates and Competitive Inhibitors of GABA Aminotransferase.” (73) Assignee: Children's Medical Center Bioorg. Med. Chem. Left., 18:3122-3125, 2008. Crewther et al., “Potassium Channel and NKCC Cotransporter Corporation, Boston, MA (US) Involvement in Ocular Refractive Control Mechanisms. PlosOne, 3:e2839, 2008. (*) Notice: Subject to any disclaimer, the term of this Cubells et al., “In vivo action of enzyme-activated irreversible inhibi patent is extended or adjusted under 35 tors of glutamic acid decarboxylase and gamma-aminobutyric acid U.S.C. 154(b) by 149 days. transaminase in retina vs. brain.” J. Pharmacol. Exp. Ther. 238:508 514, 1986 (Abstract only). (21) Appl. No.: 13/069,311 Duboc et al., “Vigabatrin, the GABA-transaminase inhibitor, dam ages cone photoreceptors in rats.” Ann. Neurol. 55:695-705, 2004 (22) Filed: Mar 22, 2011 (Abstract only). Dzhala et al., “Bumetanide enhances phenobarbital efficacy in a (65) Prior Publication Data neonatal seizure model.” Ann. Neurol. 63:222-235, 2008 (Abstract US 2011/0237554 A1 Sep. 29, 2011 only). Dzhala et al., “NKCC1 transporter facilitates seizures in the devel oping brain.” Nature Med., 11:1205-1213, 2005 (Abstract only). Follett et al., “Glutamate Receptor-Mediated Oligodendrocyte Tox Related U.S. -

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -

Thesis Rests with Its Author
University of Bath PHD A modelling study of the ligand-gated ion-channel superfamily of receptors Cockcroft, Victor Barrie Award date: 1992 Awarding institution: University of Bath Link to publication Alternative formats If you require this document in an alternative format, please contact: [email protected] General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 08. Oct. 2021 A MODELLING STUDY OF THE LIGAND-GATED ION-CHANNEL SUPERFAMILY OF RECEPTORS. submitted by Victor Barrie Cockcroft for the degree of Ph.D. at the University of Bath 1992 Copyright Attention is drawn to the fact that the copyright of this thesis rests with its author. This copy of the thesis has been supplied on the condition that anyone who con sults it is understood to recognize that its copyright rests with its author and that no quotation from the thesis and no information derived from it may be published with out the prior written consent of the author. -

The Syntheses of Β-Lactam Antibiotics from 3-Formylcephalosporins As the Key-Intermediates
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/19082 Please be advised that this information was generated on 2021-10-03 and may be subject to change. The Syntheses of β-Lactam Antibiotics from 3-Formylcephalosporins as the Key-intermediates Een wetenschappelijke proeve op het gebied van de Natuurwetenschappen, Wiskunde en Informatica Proefschrift ter verkrijging van de graad van doctor aan de Katholieke Universiteit Nijmegen, volgens besluit van het College van Decanen in het openbaar te verdedigen op woensdag 6 februari 2002 des namiddags om 1.30 uur precies door Rolf Keltjens Geboren op 9 april 1972 te Horst Promotor Prof. dr. B. Zwanenburg Copromotor Dr. A.J.H. Klunder Manuscriptcommissie Dr. E. de Vroom (DSM Anti-Infectives, Delft) Dr. J. Verweij (voorheen DSM Anti-Infectives, Delft) Prof. dr. ir. J.C.M. van Hest The research described in this PhD thesis was part of the Cluster Project "Fine- Chemistry" and was financially supported by DSM (Geleen, The Netherlands) and the Dutch Ministry of Economical Affairs (Senter). ISBN 90-9015389-6 Omslag: Penicillium chrysogenum (copyright DSM N.V.) "Experiment is the sole interpreter of the artifices of Nature" Leonardo da Vinci (1452-1519) Aan mijn ouders Paranimfen René Gieling Sander Hornes VOORWOORD Als het manuscript nagenoeg gereed is en het geheel klaar is voor verzending naar de drukker, is het ook de hoogste tijd om de mensen met wie je vier jaar lang intensief hebt samengewerkt te bedanken. -

Cell Surface Mobility of GABAB Receptors Saad Bin
Cell surface mobility of GABAB receptors Saad Bin Hannan September 2011 A thesis submitted in fulfilment of the requirements for the degree of Doctor of Philosophy of the University College London Department of Neuroscience, Physiology, and Pharmacology University College London Gower Street London WC1E 6BT UK Declaration ii ‘I, Saad Hannan confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis.' ____________________ Saad Hannan September 2011 To Ammu, Abbu, Polu Abstract ivi Abstract Type-B γ-aminobutyric acid receptors (GABABRs) are important for mediating slow inhibition in the central nervous system and the kinetics of their internalisation and lateral mobility will be a major determinant of their signalling efficacy. Functional GABABRs require R1 and R2 subunit co-assembly, but how heterodimerisation affects the trafficking kinetics of GABABRs is unknown. Here, an α- bungarotoxin binding site (BBS) was inserted into the N-terminus of R2 to monitor receptor mobility in live cells. GABABRs are internalised via clathrin- and dynamin- dependent pathways and recruited to endosomes. By mutating the BBS, a new technique was developed to differentially track R1a and R2 simultaneously, revealing the subunits internalise as heteromers and that R2 dominantly-affects constitutive internalisation of GABABRs. Notably, the internalisation profile of R1aR2 heteromers, but not R1a homomers devoid of their ER retention motif (R1ASA), is similar to R2 homomers in heterologous systems. The internalisation of R1aASA was slowed to that of R2 by mutating a di-leucine motif in the R1 C-terminus, indicating a new role for heterodimerisation, whereby R2 subunits slow the internalization of surface GABABRs. -

Design, Synthesis, and Evaluation of Antiepileptic Compounds Based on Β-Alanine and Isatin
Design, Synthesis, and Evaluation of Antiepileptic Compounds Based on β-Alanine and Isatin by Robert Philip Colaguori A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Pharmaceutical Sciences University of Toronto © Copyright by Robert Philip Colaguori, 2016 ii Design, Synthesis, and Evaluation of Antiepileptic Compounds Based on β-Alanine and Isatin Robert Philip Colaguori Master of Science Department of Pharmaceutical Sciences University of Toronto 2016 Abstract Epilepsy is the fourth-most common neurological disorder in the world. Approximately 70% of cases can be controlled with therapeutics, however 30% remain pharmacoresistant. There is no cure for the disorder, and patients affected are subsequently medicated for life. Thus, there is a need to develop compounds that can treat not only the symptoms, but also delay/prevent progression. Previous work resulted in the discovery of NC-2505, a substituted β-alanine with activity against chemically induced seizures. Several N- and α-substituted derivatives of this compound were synthesized and evaluated in the kindling model and 4-AP model of epilepsy. In the kindling model, RC1-080 and RC1-102 were able to decrease the mean seizure score from 5 to 3 in aged mice. RC1-085 decreased the interevent interval by a factor of 2 in the 4-AP model. Future studies are focused on the synthesis of further compounds to gain insight on structure necessary for activity. iii Acknowledgments First and foremost, I would like to thank my supervisor Dr. Donald Weaver for allowing me to join the lab as a graduate student and perform the work ultimately resulting in this thesis. -

1 Abietic Acid R Abrasive Silica for Polishing DR Acenaphthene M (LC
1 abietic acid R abrasive silica for polishing DR acenaphthene M (LC) acenaphthene quinone R acenaphthylene R acetal (see 1,1-diethoxyethane) acetaldehyde M (FC) acetaldehyde-d (CH3CDO) R acetaldehyde dimethyl acetal CH acetaldoxime R acetamide M (LC) acetamidinium chloride R acetamidoacrylic acid 2- NB acetamidobenzaldehyde p- R acetamidobenzenesulfonyl chloride 4- R acetamidodeoxythioglucopyranose triacetate 2- -2- -1- -β-D- 3,4,6- AB acetamidomethylthiazole 2- -4- PB acetanilide M (LC) acetazolamide R acetdimethylamide see dimethylacetamide, N,N- acethydrazide R acetic acid M (solv) acetic anhydride M (FC) acetmethylamide see methylacetamide, N- acetoacetamide R acetoacetanilide R acetoacetic acid, lithium salt R acetobromoglucose -α-D- NB acetohydroxamic acid R acetoin R acetol (hydroxyacetone) R acetonaphthalide (α)R acetone M (solv) acetone ,A.R. M (solv) acetone-d6 RM acetone cyanohydrin R acetonedicarboxylic acid ,dimethyl ester R acetonedicarboxylic acid -1,3- R acetone dimethyl acetal see dimethoxypropane 2,2- acetonitrile M (solv) acetonitrile-d3 RM acetonylacetone see hexanedione 2,5- acetonylbenzylhydroxycoumarin (3-(α- -4- R acetophenone M (LC) acetophenone oxime R acetophenone trimethylsilyl enol ether see phenyltrimethylsilyl... acetoxyacetone (oxopropyl acetate 2-) R acetoxybenzoic acid 4- DS acetoxynaphthoic acid 6- -2- R 2 acetylacetaldehyde dimethylacetal R acetylacetone (pentanedione -2,4-) M (C) acetylbenzonitrile p- R acetylbiphenyl 4- see phenylacetophenone, p- acetyl bromide M (FC) acetylbromothiophene 2- -5- -

5-HT2A/2C Receptor Modulation of Absence Seizures and Characterization of the GHB-Model
5-HT2A/2C receptor modulation of absence seizures and characterization of the GHB-model A thesis submitted to Cardiff University for the degree of Doctor of Philosophy by Marcello Venzi (MSc) School of Biosciences, Cardiff University, October 2014 Chapter 1 Declaration and Statements This work has not been submitted in substance for any other degree or award at this or any other university or place of learning, nor is being submitted concurrently in candidature for any degree or other award. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 1 This thesis is being submitted in partial fulfillment of the requirements for the degree of PhD . Signed ………………………………………… (candidate) Date ………………………… STATEMENT 2 This thesis is the result of my own independent work/investigation, except where otherwise stated. Other sources are acknowledged by explicit references. The views expressed are my own. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 3 I hereby give consent for my thesis, if accepted, to be available for photocopying and for inter- library loan, and for the title and summary to be made available to outside organisations. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 4: PREVIOUSLY APPROVED BAR ON ACCESS I hereby give consent for my thesis, if accepted, to be available for photocopying and for inter- library loans after expiry of a bar on access previously approved by the Academic Standards & Quality Committee. Signed ………………………………………… (candidate) Date ………………………… II Chapter 1 Summary Absence seizures (ASs) are non-convulsive epileptic events which are common in pediatric and juvenile epilepsies. They consist of EEG generalized spike-and-wave-discharges (SWDs) accompanied by an impairment of consciousness and are expressed within the thalamocortical network. -

Isoguvacine Hydrochloride | Medchemexpress
Inhibitors Product Data Sheet Isoguvacine hydrochloride • Agonists Cat. No.: HY-100810 CAS No.: 68547-97-7 Molecular Formula: C₆H₁₀ClNO₂ • Molecular Weight: 163.6 Screening Libraries Target: GABA Receptor Pathway: Membrane Transporter/Ion Channel; Neuronal Signaling Storage: Powder -20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month SOLVENT & SOLUBILITY In Vitro H2O : ≥ 100 mg/mL (611.25 mM) DMSO : 25 mg/mL (152.81 mM; Need ultrasonic) * "≥" means soluble, but saturation unknown. Mass Solvent 1 mg 5 mg 10 mg Concentration Preparing 1 mM 6.1125 mL 30.5623 mL 61.1247 mL Stock Solutions 5 mM 1.2225 mL 6.1125 mL 12.2249 mL 10 mM 0.6112 mL 3.0562 mL 6.1125 mL Please refer to the solubility information to select the appropriate solvent. In Vivo 1. Add each solvent one by one: 10% DMSO >> 40% PEG300 >> 5% Tween-80 >> 45% saline Solubility: ≥ 2.08 mg/mL (12.71 mM); Clear solution 2. Add each solvent one by one: 10% DMSO >> 90% (20% SBE-β-CD in saline) Solubility: ≥ 2.08 mg/mL (12.71 mM); Clear solution 3. Add each solvent one by one: 10% DMSO >> 90% corn oil Solubility: ≥ 2.08 mg/mL (12.71 mM); Clear solution BIOLOGICAL ACTIVITY Description Isoguvacine hydrochloride is a GABA receptor agonist. IC₅₀ & Target GABA[1] In Vitro Isoguvacine binds to a mouse forebrain synaptic membrane preparation. The specific binding is displaceable by GABA, Page 1 of 2 www.MedChemExpress.com muscimol and bicuculline but not by picrotoxin or diaminobutyric acid. -

Gabaergic Signaling Linked to Autophagy Enhances Host Protection Against Intracellular Bacterial Infections
ARTICLE DOI: 10.1038/s41467-018-06487-5 OPEN GABAergic signaling linked to autophagy enhances host protection against intracellular bacterial infections Jin Kyung Kim1,2,3, Yi Sak Kim1,2,3, Hye-Mi Lee1,3, Hyo Sun Jin4, Chiranjivi Neupane 2,5, Sup Kim1,2,3, Sang-Hee Lee6, Jung-Joon Min7, Miwa Sasai8, Jae-Ho Jeong 9,10, Seong-Kyu Choe11, Jin-Man Kim12, Masahiro Yamamoto8, Hyon E. Choy 9,10, Jin Bong Park 2,5 & Eun-Kyeong Jo1,2,3 1234567890():,; Gamma-aminobutyric acid (GABA) is the principal inhibitory neurotransmitter in the brain; however, the roles of GABA in antimicrobial host defenses are largely unknown. Here we demonstrate that GABAergic activation enhances antimicrobial responses against intracel- lular bacterial infection. Intracellular bacterial infection decreases GABA levels in vitro in macrophages and in vivo in sera. Treatment of macrophages with GABA or GABAergic drugs promotes autophagy activation, enhances phagosomal maturation and antimicrobial responses against mycobacterial infection. In macrophages, the GABAergic defense is mediated via macrophage type A GABA receptor (GABAAR), intracellular calcium release, and the GABA type A receptor-associated protein-like 1 (GABARAPL1; an Atg8 homolog). Finally, GABAergic inhibition increases bacterial loads in mice and zebrafish in vivo, sug- gesting that the GABAergic defense plays an essential function in metazoan host defenses. Our study identified a previously unappreciated role for GABAergic signaling in linking antibacterial autophagy to enhance host innate defense against intracellular bacterial infection. 1 Department of Microbiology, Chungnam National University School of Medicine, Daejeon 35015, Korea. 2 Department of Medical Science, Chungnam National University School of Medicine, Daejeon 35015, Korea.