"2-Oxo-1-Pyrrolidine Derivative and Its Pharmaceutical Uses"
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
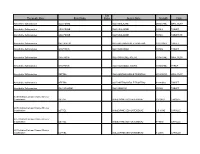
Therapeutic Class Brand Name P a Status Generic
P A Therapeutic Class Brand Name Status Generic Name Strength Form Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 250MG/5ML ORAL SUSP Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 500MG TABLET Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 500MG TABLET DR Absorbable Sulfonamides BACTRIM DS SULFAMETHOXAZOLE/TRIMETHO 800-160MG TABLET Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE 500MG TABLET Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE ACETYL 500MG/5ML ORAL SUSP Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE ACETYL 500MG/5ML SYRUP Absorbable Sulfonamides SEPTRA SULFAMETHOXAZOLE/TRIMETHO 200-40MG/5 ORAL SUSP Absorbable Sulfonamides SEPTRA SULFAMETHOXAZOLE/TRIMETHO 400-80MG TABLET Absorbable Sulfonamides SULFADIAZINE SULFADIAZINE 500MG TABLET ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 10-20MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 2.5-10MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-10MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-20MG CAPSULE P A Therapeutic Class Brand Name Status Generic Name Strength Form ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-40MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 10-40MG CAPSULE Acne Agents, Systemic ACCUTANE ISOTRETINOIN 10MG CAPSULE Acne Agents, Systemic ACCUTANE ISOTRETINOIN 20MG CAPSULE Acne Agents, Systemic ACCUTANE -

STATUS EPILEPTICUS in ADULTS (Convulsive Seizures in Patients Aged > 16 Years Old) Link Consultant: Dr Hannah Cock
STATUS EPILEPTICUS IN ADULTS (Convulsive Seizures in patients aged > 16 years old) Link consultant: Dr Hannah Cock Status epilepticus (SE) is defined as continuous seizure activity which has failed to self- terminate leading to a risk of neurological damage. The risks are highest with generalised tonic/clonic (convulsive) seizures. Convulsive SE may present as either a run of discreet generalised tonic/clonic seizures without full recovery in between (ie without regaining consciousness), or continuous generalised tonic/clonic seizure activity. Most convulsive seizures terminate spontaneously within 3 minutes, and do NOT need emergency treatment. Convulsive seizures lasting longer than 5 minutes, or recurring without recovery should be managed as Convulsive SE, unless the patient is known to habitually have longer seizures with self-termination (eg information from relatives, friends, or the patient’s epilepsy card or diary). The mortality and morbidity of generalised status epilepticus is high, and it is important to control fits as soon as possible, to use adequate doses of 1st and 2nd line agents, but not to over-treat patients in whom seizures have terminated but are slow to recover. GENERAL MANAGEMENT 1st stage (0-10mins). Protect the patient e.g. padded bed rails. Do not restrain. Administer oxygen. During an inter-ictal period insert an airway and then administer oxygen. Do not attempt to insert anything in the patient’s mouth during a seizure, even if the tongue is injured. Place the patient in a semi-prone position with the head down to prevent aspiration. Establish iv access. Note the time. 2nd Stage (0-30mins). Institute regular monitoring (temperature, cardiac, respiration, BP). -

In Silico Methods for Drug Repositioning and Drug-Drug Interaction Prediction
In silico Methods for Drug Repositioning and Drug-Drug Interaction Prediction Pathima Nusrath Hameed ORCID: 0000-0002-8118-9823 Submitted in total fulfilment of the requirements for the degree of Doctor of Philosophy Department of Mechanical Engineering THE UNIVERSITY OF MELBOURNE May 2018 Copyright © 2018 Pathima Nusrath Hameed All rights reserved. No part of the publication may be reproduced in any form by print, photoprint, microfilm or any other means without written permission from the author. Abstract Drug repositioning and drug-drug interaction (DDI) prediction are two fundamental ap- plications having a large impact on drug development and clinical care. Drug reposi- tioning aims to identify new uses for existing drugs. Moreover, understanding harmful DDIs is essential to enhance the effects of clinical care. Exploring both therapeutic uses and adverse effects of drugs or a pair of drugs have significant benefits in pharmacology. The use of computational methods to support drug repositioning and DDI prediction en- able improvements in the speed of drug development compared to in vivo and in vitro methods. This thesis investigates the consequences of employing a representative training sam- ple in achieving better performance for DDI classification. The Positive-Unlabeled Learn- ing method introduced in this thesis aims to employ representative positives as well as reliable negatives to train the binary classifier for inferring potential DDIs. Moreover, it explores the importance of a finer-grained similarity metric to represent the pairwise drug similarities. Drug repositioning can be approached by new indication detection. In this study, Anatomical Therapeutic Chemical (ATC) classification is used as the primary source to determine the indications/therapeutic uses of drugs for drug repositioning. -

Therapeutic Class Overview Anticonvulsants
Therapeutic Class Overview Anticonvulsants INTRODUCTION Epilepsy is a disease of the brain defined by any of the following (Fisher et al 2014): ○ At least 2 unprovoked (or reflex) seizures occurring > 24 hours apart; ○ 1 unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (at least 60%) after 2 unprovoked seizures, occurring over the next 10 years; ○ Diagnosis of an epilepsy syndrome. Types of seizures include generalized seizures, focal (partial) seizures, and status epilepticus (Centers for Disease Control and Prevention [CDC] 2018, Epilepsy Foundation 2016). ○ Generalized seizures affect both sides of the brain and include: . Tonic-clonic (grand mal): begin with stiffening of the limbs, followed by jerking of the limbs and face . Myoclonic: characterized by rapid, brief contractions of body muscles, usually on both sides of the body at the same time . Atonic: characterized by abrupt loss of muscle tone; they are also called drop attacks or akinetic seizures and can result in injury due to falls . Absence (petit mal): characterized by brief lapses of awareness, sometimes with staring, that begin and end abruptly; they are more common in children than adults and may be accompanied by brief myoclonic jerking of the eyelids or facial muscles, a loss of muscle tone, or automatisms. ○ Focal seizures are located in just 1 area of the brain and include: . Simple: affect a small part of the brain; can affect movement, sensations, and emotion, without a loss of consciousness . Complex: affect a larger area of the brain than simple focal seizures and the patient loses awareness; episodes typically begin with a blank stare, followed by chewing movements, picking at or fumbling with clothing, mumbling, and performing repeated unorganized movements or wandering; they may also be called “temporal lobe epilepsy” or “psychomotor epilepsy” . -

Restless Legs Syndrome in Patients with Epilepsy Under Levetiracetam Monotherapy
Original Article / Özgün Makale DODO I: 10.4274/ I: 10.4274/jtsm.xxxjtsm.69188 Journal of Turkish Sleep Medicine 2018;5:12-6 Restless Legs Syndrome in Patients with Epilepsy Under Levetiracetam Monotherapy Levetirasetam Monoterapisi Altında Epilepsi Hastalarında Huzursuz Bacak Sendromu Gülnihal Kutlu, Fatma Genç*, Yasemin Ünal, Dilek Aslan Öztürk, Abidin Erdal*, Yasemin Biçer Gömceli* Muğla Sıtkı Koçman University Faculty of Medicine, Department of Neurology, Muğla, Turkey *University of Health Sciences, Antalya Training and Research Hospital, Clinic of Neurology, Antalya, Turkey Abstract Öz Objective: Restless Legs syndrome (RLS) is a frequent neurological Amaç: Huzursuz Bacak sendromu (HBS) sık görülen bir nörolojik disease. Levetiracetam (LEV) is an effective and broad-spectrum hastalıktır. Levetirasetam (LEV) etkili ve geniş spektrumlu bir antiepileptik anticonvulsant drug. The aim of this study is to investigate the frequency ilaçtır. Bu çalışmanın amacı epilepsi tanısı ile LEV monoterapisi alan of RLS in patients diagnosed with epilepsy who took LEV monotherapy. hastalarda HBS sıklığını araştırmaktır. Materials and Methods: Two neurologists were reviewed the files of Gereç ve Yöntem: Epilepsi polikliniğinde takip edilen 1680 hastanın 1680 patients, who were followed in epilepsy outpatient clinic. One dosyası iki nörolog tarafından gözden geçirildi. En az 6 aydır LEV hundred seven patients under LEV monotherapy for at least six months monoterapisi alan 107 hasta ve 120 sağlıklı kontrol çalışmaya alındı. and 120 healthy controls were included in the study. The criteria for the International Restless Legs Syndrome Study Group were taken into HBS değerlendirmesi için Uluslararası Huzursuz Bacak Sendromu Çalışma consideration for the assessment of RLS. Grubu’nun kriterleri göz önüne alındı. Results: The mean age of patient group was 38.26±17.39 years, while Bulgular: Sağlıklı kontrollerin ortalama yaşı 39,17±16,12 yıl iken, the mean age of healthy controls was 39.17±16.12 years. -

Epilepsy & Behavior
Epilepsy & Behavior 80 (2018) 365–369 Contents lists available at ScienceDirect Epilepsy & Behavior journal homepage: www.elsevier.com/locate/yebeh Brief Communication Eslicarbazepine acetate as a replacement for levetiracetam in people with epilepsy developing behavioral adverse events Virupakshi Jalihal a, Rohit Shankar b,c,⁎, William Henley c, Mary Parrett d, Phil Tittensor e, Brendan N. McLean d, Ammad Ahmed f, Josemir W. Sander g,h,i a Ramaiah Medical College and Hospitals, Bengaluru, Karnataka 560054, India b Cornwall Partnership NHS Foundation Trust, Threemilestone Industrial Estate, Truro TR4 9LD, UK c Exeter Medical School, Knowledge Spa, Royal Cornwall Hospital, Truro, Cornwall TR1 3HD, UK d Royal Cornwall Hospital, Truro, Cornwall TR1 3LJ, UK e Royal Wolverhampton NHS Trust, UK f Bial Pharma Ltd., Admiral House, Windsor SL4 3BL, UK g NIHR University College London Hospitals Biomedical Research Centre, UCL Institute of Neurology, Queen Square, London WC1N 3BG, UK h Chalfont Centre for Epilepsy, Chalfont St Peter, Buckinghamshire SL9 0RJ, UK i Stichting Epilepsie Instellingen Nederland (SEIN), Achterweg 5, 2103 SW Heemstede, Netherlands article info abstract Article history: Background: Psychiatric and behavioral side effects (PBSEs) are a major cause of antiepileptic drug (AED) Received 13 November 2017 withdrawal. Levetiracetam (LEV) is a recognized first-line AED with good seizure outcomes but recognized Revised 16 January 2018 with PBSEs. Eslicarbazepine (ESL) is considered to function similarly to an active metabolite of the commonly Accepted 17 January 2018 used carbamazepine (CBZ). Carbamazepine is used as psychotropic medication to assist in various psychiatric Available online 5 February 2018 illnesses such as mood disorders, aggression, and anxiety. -

Mechanisms of Action of Antiepileptic Drugs
Review Mechanisms of action of antiepileptic drugs Epilepsy affects up to 1% of the general population and causes substantial disability. The management of seizures in patients with epilepsy relies heavily on antiepileptic drugs (AEDs). Phenobarbital, phenytoin, carbamazepine and valproic acid have been the primary medications used to treat epilepsy for several decades. Since 1993 several AEDs have been approved by the US FDA for use in epilepsy. The choice of the AED is based primarily on the seizure type, spectrum of clinical activity, side effect profile and patient characteristics such as age, comorbidities and concurrent medical treatments. Those AEDs with broad- spectrum activity are often found to exert an action at more than one molecular target. This article will review the proposed mechanisms of action of marketed AEDs in the US and discuss the future of AEDs in development. 1 KEYWORDS: AEDs anticonvulsant drugs antiepileptic drugs epilepsy Aaron M Cook mechanism of action seizures & Meriem K Bensalem-Owen† The therapeutic armamentarium for the treat- patients with refractory seizures. The aim of this 1UK HealthCare, 800 Rose St. H-109, ment of seizures has broadened significantly article is to discuss the past, present and future of Lexington, KY 40536-0293, USA †Author for correspondence: over the past decade [1]. Many of the newer AED pharmacology and mechanisms of action. College of Medicine, Department of anti epileptic drugs (AEDs) have clinical advan- Neurology, University of Kentucky, 800 Rose Street, Room L-455, tages over older, so-called ‘first-generation’ First-generation AEDs Lexington, KY 40536, USA AEDs in that they are more predictable in their Broadly, the mechanisms of action of AEDs can Tel.: +1 859 323 0229 Fax: +1 859 323 5943 dose–response profile and typically are associ- be categorized by their effects on the neuronal [email protected] ated with less drug–drug interactions. -

Chapter 25 Mechanisms of Action of Antiepileptic Drugs
Chapter 25 Mechanisms of action of antiepileptic drugs GRAEME J. SILLS Department of Molecular and Clinical Pharmacology, University of Liverpool _________________________________________________________________________ Introduction The serendipitous discovery of the anticonvulsant properties of phenobarbital in 1912 marked the foundation of the modern pharmacotherapy of epilepsy. The subsequent 70 years saw the introduction of phenytoin, ethosuximide, carbamazepine, sodium valproate and a range of benzodiazepines. Collectively, these compounds have come to be regarded as the ‘established’ antiepileptic drugs (AEDs). A concerted period of development of drugs for epilepsy throughout the 1980s and 1990s has resulted (to date) in 16 new agents being licensed as add-on treatment for difficult-to-control adult and/or paediatric epilepsy, with some becoming available as monotherapy for newly diagnosed patients. Together, these have become known as the ‘modern’ AEDs. Throughout this period of unprecedented drug development, there have also been considerable advances in our understanding of how antiepileptic agents exert their effects at the cellular level. AEDs are neither preventive nor curative and are employed solely as a means of controlling symptoms (i.e. suppression of seizures). Recurrent seizure activity is the manifestation of an intermittent and excessive hyperexcitability of the nervous system and, while the pharmacological minutiae of currently marketed AEDs remain to be completely unravelled, these agents essentially redress the balance between neuronal excitation and inhibition. Three major classes of mechanism are recognised: modulation of voltage-gated ion channels; enhancement of gamma-aminobutyric acid (GABA)-mediated inhibitory neurotransmission; and attenuation of glutamate-mediated excitatory neurotransmission. The principal pharmacological targets of currently available AEDs are highlighted in Table 1 and discussed further below. -

(12) Patent Application Publication (10) Pub. No.: US 2010/014.3507 A1 Gant Et Al
US 2010.0143507A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/014.3507 A1 Gant et al. (43) Pub. Date: Jun. 10, 2010 (54) CARBOXYLIC ACID INHIBITORS OF Publication Classification HISTONE DEACETYLASE, GABA (51) Int. Cl. TRANSAMINASE AND SODIUM CHANNEL A633/00 (2006.01) A 6LX 3/553 (2006.01) A 6LX 3/553 (2006.01) (75) Inventors: Thomas G. Gant, Carlsbad, CA A63L/352 (2006.01) (US); Sepehr Sarshar, Cardiff by A6II 3/19 (2006.01) the Sea, CA (US) C07C 53/128 (2006.01) A6IP 25/06 (2006.01) A6IP 25/08 (2006.01) Correspondence Address: A6IP 25/18 (2006.01) GLOBAL PATENT GROUP - APX (52) U.S. Cl. .................... 424/722:514/211.13: 514/221; 10411 Clayton Road, Suite 304 514/456; 514/557; 562/512 ST. LOUIS, MO 63131 (US) (57) ABSTRACT Assignee: AUSPEX The present invention relates to new carboxylic acid inhibi (73) tors of histone deacetylase, GABA transaminase, and/or PHARMACEUTICALS, INC., Sodium channel activity, pharmaceutical compositions Vista, CA (US) thereof, and methods of use thereof. (21) Appl. No.: 12/632,507 Formula I (22) Filed: Dec. 7, 2009 Related U.S. Application Data (60) Provisional application No. 61/121,024, filed on Dec. 9, 2008. US 2010/014.3507 A1 Jun. 10, 2010 CARBOXYLIC ACID INHIBITORS OF HISTONE DEACETYLASE, GABA TRANSAMNASE AND SODIUM CHANNEL 0001. This application claims the benefit of priority of Valproic acid U.S. provisional application No. 61/121,024, filed Dec. 9, 2008, the disclosure of which is hereby incorporated by ref 0004 Valproic acid is extensively metabolised via erence as if written herein in its entirety. -

Design, Synthesis, and Evaluation of Antiepileptic Compounds Based on Β-Alanine and Isatin
Design, Synthesis, and Evaluation of Antiepileptic Compounds Based on β-Alanine and Isatin by Robert Philip Colaguori A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Pharmaceutical Sciences University of Toronto © Copyright by Robert Philip Colaguori, 2016 ii Design, Synthesis, and Evaluation of Antiepileptic Compounds Based on β-Alanine and Isatin Robert Philip Colaguori Master of Science Department of Pharmaceutical Sciences University of Toronto 2016 Abstract Epilepsy is the fourth-most common neurological disorder in the world. Approximately 70% of cases can be controlled with therapeutics, however 30% remain pharmacoresistant. There is no cure for the disorder, and patients affected are subsequently medicated for life. Thus, there is a need to develop compounds that can treat not only the symptoms, but also delay/prevent progression. Previous work resulted in the discovery of NC-2505, a substituted β-alanine with activity against chemically induced seizures. Several N- and α-substituted derivatives of this compound were synthesized and evaluated in the kindling model and 4-AP model of epilepsy. In the kindling model, RC1-080 and RC1-102 were able to decrease the mean seizure score from 5 to 3 in aged mice. RC1-085 decreased the interevent interval by a factor of 2 in the 4-AP model. Future studies are focused on the synthesis of further compounds to gain insight on structure necessary for activity. iii Acknowledgments First and foremost, I would like to thank my supervisor Dr. Donald Weaver for allowing me to join the lab as a graduate student and perform the work ultimately resulting in this thesis. -

Mode of Seizure Inhibition by Sodium Channel Blockers, an SV2A Ligand
Epilepsy Research 154 (2019) 42–49 Contents lists available at ScienceDirect Epilepsy Research journal homepage: www.elsevier.com/locate/epilepsyres Mode of seizure inhibition by sodium channel blockers, an SV2A ligand, and T an AMPA receptor antagonist in a rat amygdala kindling model ⁎ Ting Wua, Katsutoshi Idoa, Makoto Ohgoha, Takahisa Hanadab, a Neurology Tsukuba Research Department, Discovery, Medicine Creation, Neurology Business Group, Eisai Co., Ltd. Japan b Clinical Science Department, Medical Division, Eisai Co., Ltd. Nishigokencho 13-1, Shinjuku-ku, Tokyo 162-0812, Japan ARTICLE INFO ABSTRACT Keywords: Purpose: A number of antiepileptic drugs (AEDs) with a variety of modes of action, are effective in treating focal AMPA receptor antagonist seizures. Several AEDs, such as perampanel (PER), levetiracetam (LEV), lacosamide (LCM), lamotrigine (LTG), Antiepileptic drug and carbamazepine (CBZ), have been shown to elevate the seizure threshold in kindling models. These AEDs are Mode of seizure inhibition clinically effective, but differences exist in the anti-seizure profiles of drugs with similar modes ofaction. Focal seizure Therefore, we hypothesized that there are differences in how these AEDs affect seizures. Here, we evaluated the Perampanel effects of AEDs on various seizure parameters in a rat amygdala kindling model upon stimulation attheafter- Synaptic transmission discharge threshold (ADT) and at three-times the ADT (3xADT) to characterize the differences in the effects of these AEDs. Methods: PER, LEV, LCM, LTG, CBZ, or vehicle was administered intraperitoneally to fully kindled rats. Changes in Racine seizure score, after-discharge duration (ADD), and latency to Racine score 4 generalized seizure (S4L) were measured to assess differences in the modes of seizure inhibition among the AEDs. -

High-Throughput Brain Activity Mapping and Machine Learning As a Foundation for Systems Neuropharmacology
High-throughput brain activity mapping and machine learning as a foundation for systems neuropharmacology Lin, Xudong; Duan, Xin; Jacobs, Claire; Ullmann, Jeremy; Chan, Chung-Yuen; Chen, Siya; Cheng, Shuk-Han; Zhao, Wen-Ning; Poduri, Annapurna; Wang, Xin; Haggarty, Stephen J.; Shi, Peng Published in: Nature Communications Published: 03/12/2018 Document Version: Final Published version, also known as Publisher’s PDF, Publisher’s Final version or Version of Record License: CC BY Publication record in CityU Scholars: Go to record Published version (DOI): 10.1038/s41467-018-07289-5 Publication details: Lin, X., Duan, X., Jacobs, C., Ullmann, J., Chan, C-Y., Chen, S., Cheng, S-H., Zhao, W-N., Poduri, A., Wang, X., Haggarty, S. J., & Shi, P. (2018). High-throughput brain activity mapping and machine learning as a foundation for systems neuropharmacology. Nature Communications, 9, [5142]. https://doi.org/10.1038/s41467-018-07289- 5 Citing this paper Please note that where the full-text provided on CityU Scholars is the Post-print version (also known as Accepted Author Manuscript, Peer-reviewed or Author Final version), it may differ from the Final Published version. When citing, ensure that you check and use the publisher's definitive version for pagination and other details. General rights Copyright for the publications made accessible via the CityU Scholars portal is retained by the author(s) and/or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Users may not further distribute the material or use it for any profit-making activity or commercial gain.