A Web Tool for the Design of Prime-Editing Guide Rnas
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
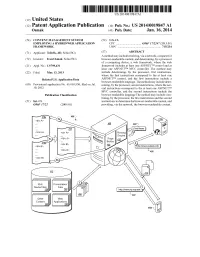
(12) Patent Application Publication (10) Pub. No.: US 2014/0019847 A1 Osmak (43) Pub
US 20140019847A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0019847 A1 OSmak (43) Pub. Date: Jan. 16, 2014 (54) CONTENT MANAGEMENT SYSTEM (52) U.S. Cl. EMPLOYINGA HYBRD WEB APPLICATION CPC .................................. G06F 17/2247 (2013.01) FRAMEWORK USPC .......................................................... 71.5/234 (71) Applicant: Telerik, AD, Sofia (BG) (57) ABSTRACT A method may include receiving, via a network, a request for (72) Inventor: Ivan Osmak, Sofia (BG) browser-renderable content, and determining, by a processor of a computing device, a web framework, where the web (21) Appl. No.: 13/799,431 framework includes at least one ASP.NETTM control and at least one ASP.NETTM MVC controller. The method may (22) Filed: Mar 13, 2013 include determining, by the processor, first instructions, where the first instructions correspond to the at least one Related U.S. Application Data ASP.NETTM control, and the first instructions include a browser-renderable language. The method may include deter (60) Provisional application No. 61/669,930, filed on Jul. mining, by the processor, second instructions, where the sec 10, 2012. ond instructions correspond to the at least one ASP.NETTM MVC controller, and the second instructions include the Publication Classification browser-renderable language The method may include com bining, by the processor, the first instructions and the second (51) Int. Cl. instructions to determine the browser-renderable content, and G06F 7/22 (2006.01) providing, via the network, the browser-renderable content. Routing Engine Ric Presentation Media Fies : Fies 22 Applications 28 Patent Application Publication Jan. 16, 2014 Sheet 1 of 8 US 2014/001.9847 A1 Patent Application Publication Jan. -

Bachelorarbeit
BACHELORARBEIT Realisierung von verzögerungsfreien Mehrbenutzer Webapplikationen auf Basis von HTML5 WebSockets Hochschule Harz University of Applied Sciences Wernigerode Fachbereich Automatisierung und Informatik im Fach Medieninformatik Erstprüfer: Prof. Jürgen K. Singer, Ph.D. Zweitprüfer: Prof. Dr. Olaf Drögehorn Erstellt von: Lars Häuser Datum: 16.06.2011 Einleitung Inhaltsverzeichnis 1 Einleitung ................................................................................................................. 5 1.1 Zielsetzung ..................................................................................................... 5 1.2 Aufbau der Arbeit ........................................................................................... 6 2 Grundlagen .............................................................................................................. 8 2.1 TCP/IP ............................................................................................................ 8 2.2 HTTP .............................................................................................................. 9 2.3 Request-Response-Paradigma (HTTP-Request-Cycle) .............................. 10 2.4 Klassische Webanwendung: Synchrone Datenübertragung ....................... 11 2.5 Asynchrone Webapplikationen .................................................................... 11 2.6 HTML5 ......................................................................................................... 12 3 HTML5 WebSockets ............................................................................................. -

Pragmaticperl-Interviews-A4.Pdf
Pragmatic Perl Interviews pragmaticperl.com 2013—2015 Editor and interviewer: Viacheslav Tykhanovskyi Covers: Marko Ivanyk Revision: 2018-03-02 11:22 © Pragmatic Perl Contents 1 Preface .......................................... 1 2 Alexis Sukrieh (April 2013) ............................... 2 3 Sawyer X (May 2013) .................................. 10 4 Stevan Little (September 2013) ............................. 17 5 chromatic (October 2013) ................................ 22 6 Marc Lehmann (November 2013) ............................ 29 7 Tokuhiro Matsuno (January 2014) ........................... 46 8 Randal Schwartz (February 2014) ........................... 53 9 Christian Walde (May 2014) .............................. 56 10 Florian Ragwitz (rafl) (June 2014) ........................... 62 11 Curtis “Ovid” Poe (September 2014) .......................... 70 12 Leon Timmermans (October 2014) ........................... 77 13 Olaf Alders (December 2014) .............................. 81 14 Ricardo Signes (January 2015) ............................. 87 15 Neil Bowers (February 2015) .............................. 94 16 Renée Bäcker (June 2015) ................................ 102 17 David Golden (July 2015) ................................ 109 18 Philippe Bruhat (Book) (August 2015) . 115 19 Author .......................................... 123 i Preface 1 Preface Hello there! You have downloaded a compilation of interviews done with Perl pro- grammers in Pragmatic Perl journal from 2013 to 2015. Since the journal itself is in Russian -

What Is Perl
AdvancedAdvanced PerlPerl TechniquesTechniques DayDay 22 Dave Cross Magnum Solutions Ltd [email protected] Schedule 09:45 – Begin 11:15 – Coffee break (15 mins) 13:00 – Lunch (60 mins) 14:00 – Begin 15:30 – Coffee break (15 mins) 17:00 – End FlossUK 24th February 2012 Resources Slides available on-line − http://mag-sol.com/train/public/2012-02/ukuug Also see Slideshare − http://www.slideshare.net/davorg/slideshows Get Satisfaction − http://getsatisfaction.com/magnum FlossUK 24th February 2012 What We Will Cover Modern Core Perl − What's new in Perl 5.10, 5.12 & 5.14 Advanced Testing Database access with DBIx::Class Handling Exceptions FlossUK 24th February 2012 What We Will Cover Profiling and Benchmarking Object oriented programming with Moose MVC Frameworks − Catalyst PSGI and Plack FlossUK 24th February 2012 BenchmarkingBenchmarking && ProfilingProfiling Benchmarking Ensure that your program is fast enough But how fast is fast enough? premature optimization is the root of all evil − Donald Knuth − paraphrasing Tony Hoare Don't optimise until you know what to optimise FlossUK 24th February 2012 Benchmark.pm Standard Perl module for benchmarking Simple usage use Benchmark; my %methods = ( method1 => sub { ... }, method2 => sub { ... }, ); timethese(10_000, \%methods); Times 10,000 iterations of each method FlossUK 24th February 2012 Benchmark.pm Output Benchmark: timing 10000 iterations of method1, method2... method1: 6 wallclock secs \ ( 2.12 usr + 3.47 sys = 5.59 CPU) \ @ 1788.91/s (n=10000) method2: 3 wallclock secs \ ( 0.85 usr + 1.70 sys = 2.55 CPU) \ @ 3921.57/s (n=10000) FlossUK 24th February 2012 Timed Benchmarks Passing timethese a positive number runs each piece of code a certain number of times Passing timethese a negative number runs each piece of code for a certain number of seconds FlossUK 24th February 2012 Timed Benchmarks use Benchmark; my %methods = ( method1 => sub { .. -

Modern Perl, Fourth Edition
Prepared exclusively for none ofyourbusiness Prepared exclusively for none ofyourbusiness Early Praise for Modern Perl, Fourth Edition A dozen years ago I was sure I knew what Perl looked like: unreadable and obscure. chromatic showed me beautiful, structured expressive code then. He’s the right guy to teach Modern Perl. He was writing it before it existed. ➤ Daniel Steinberg President, DimSumThinking, Inc. A tour de force of idiomatic code, Modern Perl teaches you not just “how” but also “why.” ➤ David Farrell Editor, PerlTricks.com If I had to pick a single book to teach Perl 5, this is the one I’d choose. As I read it, I was reminded of the first time I read K&R. It will teach everything that one needs to know to write Perl 5 well. ➤ David Golden Member, Perl 5 Porters, Autopragmatic, LLC I’m about to teach a new hire Perl using the first edition of Modern Perl. I’d much rather use the updated copy! ➤ Belden Lyman Principal Software Engineer, MediaMath It’s not the Perl book you deserve. It’s the Perl book you need. ➤ Gizmo Mathboy Co-founder, Greater Lafayette Open Source Symposium (GLOSSY) Prepared exclusively for none ofyourbusiness We've left this page blank to make the page numbers the same in the electronic and paper books. We tried just leaving it out, but then people wrote us to ask about the missing pages. Anyway, Eddy the Gerbil wanted to say “hello.” Prepared exclusively for none ofyourbusiness Modern Perl, Fourth Edition chromatic The Pragmatic Bookshelf Dallas, Texas • Raleigh, North Carolina Prepared exclusively for none ofyourbusiness Many of the designations used by manufacturers and sellers to distinguish their products are claimed as trademarks. -

Mojolicious Web Clients Brian D Foy Mojolicious Web Clients by Brian D Foy
Mojolicious Web Clients brian d foy Mojolicious Web Clients by brian d foy Copyright 2019-2020 © brian d foy. All rights reserved. Published by Perl School. ii | Table of contents Preface vii What You Should Already Know . viii Some eBook Notes ........................... ix Installing Mojolicious ......................... x Getting Help .............................. xii Acknowledgments . xiii Perl School ............................... xiv Changes ................................. xiv 1 Introduction 1 The Mojo Philosophy .......................... 1 Be Nice to Servers ........................... 4 How HTTP Works ........................... 6 Add to the Request ........................... 15 httpbin ................................. 16 Summary ................................ 18 2 Some Perl Features 19 Perl Program Basics .......................... 19 Declaring the Version ......................... 20 Signatures ................................ 22 Postfix Dereference ........................... 25 Indented Here Docs ........................... 25 Substitution Returns the Modified Copy . 26 Summary ................................ 27 3 Basic Utilities 29 Working with URLs .......................... 30 Decoding JSON ............................. 34 Collections ............................... 45 Random Utilities ............................ 50 TABLE OF CONTENTS | iii Events .................................. 52 Summary ................................ 55 4 The Document Object Model 57 Walking Through HTML or XML ................... 57 Modifying -

[email protected] 323-327
[email protected] ● http://ownlifeful.com ● 323-327-7830 ● Senior Software Engineer with 23+ years of web programming expertise. TECHNICAL SKILLS Languages: Perl, JavaScript, Python, Ruby, SQL. Environments: Linux ( Red Hat, Ubuntu ), UNIX ( macOS, FreeBSD ). Databases: MySQL, MongoDB, Oracle, PostgreSQL, Sybase, SQLite. Miscellaneous: Node.JS, Apache, REST, git, GitHub, Jira, jQuery, Bootstrap, Rails, React, Angular, HTML, CSS. RECENT EXPERIENCE Medical Leave 2019-2020 ● Took time off to undergo major heart surgery. Now recovered. Greystar Solutions, Remote 2018 - 2018 Cryptocurrency Web Applications Programmer ● Used Perl, Mojolicious, Postgres Pub/Sub, web sockets, JavaScript, Highcharts, and the Coinbase API, to create near-real-time analytics charts from live cryptocurrency data. SAP Ariba, Palo Alto, CA 2018 - 2018 DevOps Engineer ● Responsible for maintaining and enhancing a set of Perl and Python scripts used for building, deploying & monitoring enterprise applications. Sabbatical, Bonsall, CA 2016 - 2018 ● Developed a Graph Theory algorithm implemented as a Perl module. Capital One, San Francisco, CA 2015 – 2016 Senior Software Engineer ( remote ) ● Developed a B2B mobile and web-facing application to support merchants’ ability to process credit card payments. ● Created integrations with third-party REST API’s, such as Lexis Nexis. ● Enhanced the RESTful, MVC application which used Perl, Dancer, DBIx::Class, Template Toolkit, jQuery, AngularJS, Bootstrap, & MySQL. ● Wrote Test::More unit tests, and participated in a code review process. ● Participated in a distributed, Agile software team environment. NetApp, Sunnyvale, CA 2014 – 2015 Consultant ● Wrote an ETL application in Perl, with a MySQL back-end. ● Helped implement a Continuous-Integration process using Jenkins. Rent.com, Santa Monica, CA 2013 – 2013 Consultant ● Worked on Rent.com’s Object-Oriented Perl-based web site in an Agile environment. -

Mojolicious Marcos Rebelo ([email protected]) Mojolicious
Mojolicious Marcos Rebelo ([email protected]) Mojolicious ● An amazing real-time web framework supporting a simplified single file mode through Mojolicious::Lite. ● Very clean, portable and Object Oriented pure Perl API without any hidden magic and no requirements besides Perl 5.10.1 (although 5.12+ is recommended, and optional CPAN modules will be used to provide advanced functionality if they are installed). Mojolicious ● Full stack HTTP 1.1 and WebSocket client/server implementation with IPv6, TLS, Bonjour, IDNA, Comet (long polling), chunking and multipart support. ● Built-in non-blocking I/O web server supporting libev and hot deployment, perfect for embedding. ● Automatic CGI and PSGI detection. ● JSON and HTML5/XML parser with CSS3 selector support. Marcos Rebelo ● 10 years Perl Developer ● Test-driven development fan ● Mojolicious experience: ○ I'm not a Mojolicious developer. ○ A group of JSON Back-ends ○ Short effort on the Front-end Any intelligent fool can make things bigger, more complex, and more violent. It takes a touch of genius -- and a lot of courage -- to move in the opposite direction. Albert Einstein Installation $ sudo cpan Mojolicious Hello World use Mojolicious::Lite; get '/' => sub { shift->render(text => 'Hello World!') }; app->start; ● $ hello.pl daemon Server available at http://127.0.0.1: 3000. ● $ curl http://127.0.0.1:3000/ Hello World! Generator ● There is a helper command to generate a small example application. You may generate multiple things, but two are very interesting. ● $ mojo generate app Generate -

Using Docker to Assist Q&A Forum Users
1 Using Docker to Assist Q&A Forum Users Luis Melo, Igor Wiese and Marcelo d’Amorim Abstract—Q&A forums are today a valuable tool to assist developers in programming tasks. Unfortunately, contributions to these forums are often unclear and incomplete. Docker is a container solution that enables software developers to encapsulate an operating environment and could help address reproducibility issues. This paper reports on a feasibility study to evaluate if Docker can help improve reproducibility in Stack Overflow. We started surveying Stack Overflow users to understand their perceptions on the proposal of using Docker to reproduce Stack Overflow posts. Participants were critical and mentioned two important aspects: cost and need. To validate their criticism, we conducted an exploratory study focused on understanding how costly the task of creating containers for posts is for developers. Overall, results indicate that the cost of creating containers is not high, especially due to the fact that dockerfiles are highly similar and small. Based on these findings we developed a tool, dubbed FRISK, to assist developers in creating containers for those posts. We then conducted a user study to evaluate interest of Stack Overflow developers on the tool. We found that, on average, users spent nearly ten minutes interacting with FRISK and that 45.3% of the 563 FRISK sessions we created for existing posts resulted in a successful access to the corresponding web service by the owners of the post. Overall, this paper provides early evidence that the use of Docker in Q&A forums should be encouraged for configuration-related posts. -

Resume [email protected]>, @Neil H Watson
Neil H. Watson Linux Administrator, Architect, and Developer June 17, 2016 Greater Toronto Area, <resume [email protected]>, https://github.com/neilhwatson, http://watson-wilson.ca, http://www.linkedin.com/in/neilhwatson, @neil h watson - 20 years of Linux administration experience. - Experienced in all major Linux flavours. - Experienced in large physical and virtual environments with 10,000+ hosts. Experience Independent IT Contractor October 2007 - Present - Contracting and consulting for various companies including some listed below. - Architect and builder of open source security solutions EFL, Delta Reporting, and Delta Hardening. - Configuration management with Ansible, Chef, Puppet, and CFEngine. - AWS provisioning using Terraform. - Automated testing with Travis-CI and Jenkins. - IPv6 testing and deployment. - Train and consult on systems administration, infrastructure, and security. - Skills: Ruby, Perl, Git, Test Kitchen, Hadoop, Atlassian, Serverspec, Spacewalk, Kickstart Surfeasy April 2016 - June 2016 Sr. DevOps - Helped double Surfeasy’s private cloud in just three months. - Achieved a 4x reduction in monitoring alarms. - Skills: Ruby, Ansible, Puppet, Logstash, KVM, Jira, Git, Ubuntu, Nagios Next Pathway Jan 2016 - April 2016 DevOps Consultant (contract) - Evaluating and implementing continuous integration tools to deliver Hadoop applications to Next Pathway’s banking clients. - Skills: Ansible, Perl, shell, Atlassian suite, Chef, Test Kitchen AMD June 2012 - September 2015 Sr. Linux Consultant (contract) - Designed, built, and managed automatic provisioning and management tools for 10,000+ servers. - Designed isolated host environments to meet US export laws. - Trainer and technical writer. - Skills: Perl, Shell, Git, Ruby, Serverspec, Nagios, Tomcat Debian, Red Hat Satellite, Ubuntu, RedHat Linux, Kickstart, VMware CFEngine June 2011 - April 2012 Implementation Specialist (contract) - Designed and built a 30,000 host CFEngine service supporting Linux, AIX, and Solaris in a large international bank. -

Perl by Example, Third Edition—This Book Is a Superb, Well-Written Programming Book
Praise for Ellie Quigley’s Books “I picked up a copy of JavaScript by Example over the weekend and wanted to thank you for putting out a book that makes JavaScript easy to understand. I’ve been a developer for several years now and JS has always been the ‘monster under the bed,’ so to speak. Your book has answered a lot of questions I’ve had about the inner workings of JS but was afraid to ask. Now all I need is a book that covers Ajax and Coldfusion. Thanks again for putting together an outstanding book.” —Chris Gomez, Web services manager, Zunch Worldwide, Inc. “I have been reading your UNIX® Shells by Example book, and I must say, it is brilliant. Most other books do not cover all the shells, and when you have to constantly work in an organization that uses tcsh, bash, and korn, it can become very difficult. However, your book has been indispensable to me in learning the various shells and the differences between them…so I thought I’d email you, just to let you know what a great job you have done!” —Farogh-Ahmed Usmani, B.Sc. (Honors), M.Sc., DIC, project consultant (Billing Solutions), Comverse “I have been learning Perl for about two months now; I have a little shell scripting experience but that is it. I first started withLearning Perl by O’Reilly. Good book but lacking on the examples. I then went to Programming Perl by Larry Wall, a great book for intermediate to advanced, didn’t help me much beginning Perl. -

Building an API with Mojolicious
Building an API with Mojolicious Florent Mara <[email protected]> Context ● ‘Incoming XML payload from the interweb’ ● Node.js API ● No pre-existing web framework ● Isolation required ○ jump host ○ inline credentials ○ no access to other subsystems ● Looking for a blueprint Why not extend existing Node.js API? ● It is performant but … ● High TCO (module/AWS/security … ) ● No authentication ● No tests ● Currently internal ● Not isolated ● Not conducive to feature scaling or code complexity ● Limited in-house skills ● … but it is performant Why Mojolicious? ● The website looks good ● It says it is modern ● Many blog posts document success using Mojolicious ● Documentation is clear and indicates a planned progression from Mojolicious::Lite to Mojolicious ● The tutorial was simple and worked flawlessly ● It is Perl ● Likely tests will be possible, potentially even easy Why not Dancer? Catalyst? Other Web Framework? ● Dunno Getting started ● $ cpanm -i Mojolicious ● $ mojo generate lite_app myapp.pl What happened? ● One file was created ● Uses packaged Mojolicious::Lite ● Sets plugin PODRenderer ● Sets an HTTP get method on path ‘/’ that renders a template ● Calls app->start ● Defines the template Great ... ● … for rapid prototyping ● Limited maintainability / reusability / extensibility ● Code and template within file. ● No MVC Mojolicious ● $ mojo generate app my_app.pl Our Mojolicious App (1/5) ● /lib and /t common to all projects -> top level ● /script -> /Job ● /Controller and /templates relocated ● No use for /public ● Config file is ‘somewhere up’ ● .pl is ‘somewhere upper’ Our Mojolicious App: api.pm (2/5) Our Mojolicious App: Initialization (3/5) Our Mojolicious App: Config file (4/5) Our Mojolicious App: Routes (5/5) Some testing ● use Test::Mojo; For more information http://mojolicious.org/.