Induction of Atrial Endothelial Senescence by Angiotensin II And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ryerson University Spring Graduates
Ryerson University Spring Graduates June 2020 Faculty of Arts 2 Faculty of Communication & Design 11 Faculty of Community Services 21 Faculty of Engineering and Architectural Science 35 Faculty of Science 46 Ted Rogers School of Management 54 Yeates School of Graduate Studies 71 The G. Raymond Chang School of Continuing Education 73 Faculty of Arts Pamela Sugiman Dean Faculty of Arts Janice Fukakusa Chancellor Mohamed Lachemi President and Vice-Chancellor Charmaine Hack Registrar Ryerson Gold Medal Presented to Mayah Obadia Geographic Analysis 2 Faculty of Arts Undergraduate Degree Programs Arts and Contemporary Studies Bachelor of Arts (Honours) *Diana Abo Harmouch Carmen Jajjo *Megumi Noteboom *Sima Rebecca Abrams Leya Jasat Valentina Padure Qeyam Amiri Sophie Johnson *Naiomi Marcia Perera Brodie Barrick Babina Kamalanathan Charlotte Jane Prokopec Rebecca Claire Chen Caroline Susan Kewley Regan Reynolds Erin Tanya Clarke Jessica Laurenza Joshua Ricci *Megan Lisa Devoe Claire Lowenstein Kaitlin Anganie Seepersaud *Manpreet Kaur Dhaliwal *Avigayil Margolis Gabriela Skwarko Tatum Lynn Donovan Sara McArthur Julia Macey Sullivan Faith Raha Giahi *Nadia Celeste McNairn *Helen Gillian Webb Meagan Gove *Mahbod Mehrvarz *Michael Worbanski Salem Habtom Andrew Moon Smyrna Wright *William Hanchar *Liana Gabriella Mortin Calum Jacques Potoula Mozas Criminology Bachelor of Arts (Honours) *Annabelle Adjei *Jenna Anne Giannini Veronica Hiu Lam Lee Stanislav Babinets Albina Glatman Karishma Catherine Lutchman Hela Bakhtari Farah Khaled Gregni Simbiat -

FY19 Annual Report View Report
Annual Report 2018–19 3 Introduction 5 Metropolitan Opera Board of Directors 6 Season Repertory and Events 14 Artist Roster 16 The Financial Results 20 Our Patrons On the cover: Yannick Nézet-Séguin takes a bow after his first official performance as Jeanette Lerman-Neubauer Music Director PHOTO: JONATHAN TICHLER / MET OPERA 2 Introduction The 2018–19 season was a historic one for the Metropolitan Opera. Not only did the company present more than 200 exiting performances, but we also welcomed Yannick Nézet-Séguin as the Met’s new Jeanette Lerman- Neubauer Music Director. Maestro Nézet-Séguin is only the third conductor to hold the title of Music Director since the company’s founding in 1883. I am also happy to report that the 2018–19 season marked the fifth year running in which the company’s finances were balanced or very nearly so, as we recorded a very small deficit of less than 1% of expenses. The season opened with the premiere of a new staging of Saint-Saëns’s epic Samson et Dalila and also included three other new productions, as well as three exhilarating full cycles of Wagner’s Ring and a full slate of 18 revivals. The Live in HD series of cinema transmissions brought opera to audiences around the world for the 13th season, with ten broadcasts reaching more than two million people. Combined earned revenue for the Met (box office, media, and presentations) totaled $121 million. As in past seasons, total paid attendance for the season in the opera house was 75%. The new productions in the 2018–19 season were the work of three distinguished directors, two having had previous successes at the Met and one making his company debut. -
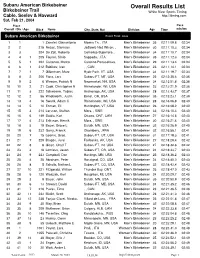
Overall Results List
Subaru American Birkebeiner Overall Results List Birkebeiner Trail White River Sports Timing Cable, Seeley & Hayward http://itiming.com Sat, Feb 21, 2004 Pl ace Pace Overall / Div / Age Bib # Name City, State, Nat Division Age Time min/km Subaru American Birkebeiner Event Field: 3833 1 1 1 Zanetel, Gianantonio Moena Tn, , ITA Men's Birkebeiner 35 02:11:09.8 02:34 2 2 215 Rezac, Stanislav Jadlowic Nad Wison, , Men's Birkebeiner 30 02:11:10.3 02:34 3 3 204 De Zolt, Roberto CoZmEelico Superiore, , Men's Birkebeiner 34 02:11:10.7 02:34 4 4 1 205 Fauner, Silvio SITaAppada, , ITA Men's Birkebeiner 35 02:11:12.3 02:34 5 5 1 202 Cattaneo, Marco Caronno Pertusellava, , Men's Birkebeiner 29 02:11:13.4 02:34 6 6 1 212 Babikov, Ivan ,I T, ACAN Men's Birkebeiner 23 02:11:14.1 02:34 7 7 1 7 Gilbertson, Marc Hyde Park, VT, USA Men's Birkebeiner 34 02:11:19.7 02:34 8 8 2 206 Flora, Lars Subaru FT, MT, USA Men's Birkebeiner 26 02:12:35.4 02:35 9 9 2 6 Weaver, Patrick N Newmarket, NH, USA Men's Birkebeiner 34 02:12:51.8 02:36 10 10 2 21 Cook, Christopher R Rhinelander, WI, USA Men's Birkebeiner 23 02:13:21.9 02:36 11 11 3 222 Schwoerer, Tobias Anchorage, AK, USA Men's Birkebeiner 28 02:13:43.7 02:37 12 12 2 56 Wadsworth, Justin Bend , OR, USA Men's Birkebeiner 35 02:15:23.1 02:39 13 13 4 16 Swank, Adam C Rhinelander, WI, USA Men's Birkebeiner 28 02:16:06.8 02:40 14 14 5 10 Enman, Eli Huntington, VT, USA Men's Birkebeiner 26 02:16:08.2 02:40 15 15 3 214 Larsson, Staffan Mora, , SWE Men's Birkebeiner 33 02:16:10.4 02:40 16 16 6 149 Saidla, Karl Ottawa, ONT, -

Your Passion for GIVING... a Foundation of HOPE
Your Passion for GIVING... A Foundation of HOPE 2012 annual report to donors and friends Dear Friends, On behalf of all the patients and families who turn to St. John Providence Health System (SJPHS) for their health care needs, thank you from the bottom of our hearts for your passion for giving. Your generous support is a foundation of hope, helping us to transform health care and provide the highest quality medical services. As we look back at the gifts received in 2012, we are amazed by the incredible generosity of our donors. Our longtime loyal supporters, new donors, grateful patients and families, corporations, foundations and organizations as well as our own associates and physicians all play an important role in total philanthropic support from the community. From the smallest, heartfelt gifts and attendance at fundraising events to the Table of Contents largest gifts supporting the expansion of our facilities and the newest technology, we thank you. Many donors, including Mike and Nancy Timmis, are drawn to support SJPHS expressly because of our Susan E. Burns Catholic health ministry and our mission to provide spiritually centered, holistic care to the community – especially the poor and vulnerable. Others, such as the Van Elslander family, have personal experiences Changing Health Care Together ................................ 2 that motivate them to support an area close to their hearts – in this case a new NICU. Then there is the remarkable generosity of the dedicated community members who wish to remain Living Our Mission .................................................... 6 anonymous in their gift to expand one of our Emergency Departments so we can better serve the growing number of patients. -

UPDATED: 12 June 2008
UPDATED: 3 December 2009 CASE-LAW REFERENCES OF JUDGMENTS AND PUBLISHED DECISIONS - A - A. and E. Riis v. Norway (no. 2), no. 16468/05, § …, 17 January 2008 A. and E. Riis v. Norway, no. 9042/04, § …, 31 May 2007 A. and Others v. Denmark, 8 February 1996, § …, Reports of Judgments and Decisions 1996-I A. and Others v. the United Kingdom [GC], no. 3455/05, § …, ECHR 2009-… A. and Others v. Turkey, no. 30015/96, § …, 27 July 2004 A. E. v. Poland, no. 14480/04, § …, 31 March 2009 A. v. France, 23 November 1993, § …, Series A no. 277-B A. v. Italy (friendly settlement), no. 40453/98, § …, 9 October 2003 A. v. Norway, no. 28070/06, § …, 9 April 2009 A. v. the United Kingdom, 23 September 1998, § …, Reports of Judgments and Decisions 1998-VI A. v. the United Kingdom, no. 35373/97, § …, ECHR 2002-X A. Yılmaz v. Turkey, no. 10512/02, § …, 22 July 2008 A.A.U. v. France, no. 44451/98, § …, 19 June 2001 A.B. v. Italy, no. 41809/98, § …, 8 February 2000 A.B. v. Poland, no. 33878/96, § …, 20 November 2007 2 A.B. v. Slovakia, no. 41784/98, § …, 4 March 2003 A.B. v. the Netherlands, no. 37328/97, § …, 29 January 2002 A.C. v. Italy, no. 44481/98, § …, 1 March 2001 A.D. v. Turkey, no. 29986/96, § …, 22 December 2005 A.D.T. v. the United Kingdom, no. 35765/97, § …, ECHR 2000-IX A.G. v. Italy, no. 66441/01, § …, 9 October 2003 A.H. v. Finland, no. 46602/99, § …, 10 May 2007 A.J. -

Public Accounts of the Province of Manitoba for the Year Ended 31St March, 1960
0 1620 0749 0426 i , ■ _ ' * PUBLIC ACCOUNTS OF THE PROVINCE OF MANITOBA FOR THE YEAR ENDED 31st MARCH, 1960 PROVINCE OF MANITOBA for the Province of Manitoba, 1960 EG GOV DOC leferenc? CAE MA F P71- 1960 ken from ta¬ bard Ex LIBRIS UNiyERSITATIS albertensis PUBLIC ACCOUNTS OF THE PROVINCE OF MANITOBA FOR THE YEAR ENDED 31st MARCH, 1960 Printed by R. S. Evans, Queen’s Printer for the Province of Manitoba, 1960 WINNIPEG Un BRARY • a rta To the Honourable Errick F. Willis, Lieutenant-Governor of the Province of Manitoba. May It Please Your Honour: The undersigned has the honour to present the Public Accounts of the Province of Manitoba for the year ended 3 1st March, 1960. DUFF ROBLIN, Acting Provincial Treasurer. Office of the Provincial Treasurer. 8th December, 1960. I E | a V ' m The Honourable Dufferin Roblin, Acting Provincial Treasurer of Manitoba. Sir: I have the honour to submit herewith the Public Accounts of the Province of Manitoba for the year ended 31st March, 1960. I have the honour to be, Sir, Your obedient servant, GEO. D. ILIFFE, F.C.A., Comptroller-General Winnipeg, Manitoba, 8th December, 1960. Public Accounts 1959-1960 7 GOVERNMENT OF THE PROVINCE OF MANITOBA ORDER OF THE PUBLIC ACCOUNTS Page Main Statements: Balance Sheet as at 31st March, 1960 . 10 Schedules to Balance Sheet as at 31st March, 1960 . 12 Statement of Revenue and Expenditure for the fiscal year ended 3'lst March, 1960 . 26 Statement of Special Warrants issued during the fiscal year ended 31st March, 1960 . 30 Comparative Statement of Revenue, 1952-4960 . -

FY17 Annual Report View Report
Annual Report 2016–17 1 2 4 Introduction 6 Metropolitan Opera Board of Directors 7 Season Repertory and Events 14 Artist Roster 16 The Financial Results 48 Our Patrons 3 Introduction In the 2016–17 season, the Metropolitan Opera continued to present outstanding grand opera, featuring the world’s finest artists, while maintaining balanced financial results—the third year running in which the company’s finances were balanced or very nearly so. The season opened with the premiere of a new production of Wagner’s Tristan und Isolde and also included five other new stagings, as well as 20 revivals. The Live in HD series of cinema transmissions brought opera to audiences around the world for the 11th year, with ten broadcasts reaching approximately 2.3 million people. Combined earned revenue for the Met (Live in HD and box office) totaled $111 million. Total paid attendance for the season in the opera house was 75%. All six new productions in the 2016–17 season were the work of distinguished directors who had previous success at the Met. The compelling Opening Night new production of Tristan und Isolde was directed by Mariusz Treliński, who made his Met debut in 2015 with the double bill of Tchaikovsky’s Iolanta and Bartók’s Bluebeard’s Castle. French-Lebanese director Pierre Audi brought his distinctive vision to Rossini’s final operatic masterpiece Guillaume Tell, following his earlier staging of Verdi’s Attila in 2010. Robert Carsen, who first worked at the Met in 1997 on his popular production of Tchaikovsky’s Eugene Onegin, directed a riveting new Der Rosenkavalier, the company’s first new staging of Strauss’s grand comedy since 1969. -

Anti-Gay Marriage Hearing Draws Hundreds to Wausau
FREE VOLUME TEN, NO. 6—March 13, 1997—March 26,1997—Issue 226 Give the People Light and they will find their own way. The Wisconsin Light Fiction, Interviews and More Debut in Anti-Gay Marriage Hearing The Wisconsin Light The Wisconsin Light is proud to an- Draws Hundreds to Wausau nounce that beginning with this issue, it will be bringing its readers several new features. Our own nationally known writer, Dr. Gays Outnumber Terry Boughner agreed to bring our readers a fabulous new story "Statues of -' Bill's Supporters At the Sacred Band" a thrilling Gay new se- rial. This action packed story features Meeting political intrigue in high places, murder, lust and love. By Bill Meunier Dr. Boughner is well known for his Exclusive On the Scene Coverage for published works including Out of All Time and numerous articles written for Les/Bi/Gay Wisconsin national Gay magazines. This exciting Wausau,WI-- Hundreds of people most serial begins in this issue with the first of them Gays, Bisexuals, Lesbians and Every is- their allies turned out for a hearing on an installment on our front page. anti sue of The Wisconsin Light will carry -Gay Marriage proposal sponsored by thriller. Fu- State Representative Lorriane Seratti, (R- another installment of this Spread gle). ture installments will be carried on the The hearing called by the our SpotLight entertain- Chidren and Family Services Committee first page of was held at the North Central Technical ment section. College Another new feature, "Dusty's Corner," Campus in Wausau on Monday "Dusty's March 10, 1997. -

Granulocyte-Colony Stimulating Factor Therapy to Induce Neovascularization in Ischemic Heart Disease
Rasmus Sejersten Granulocyte‐Colony Stimulating Factor Ripa Therapy to Induce Neovascularization Granulocyte in Ischemic Heart Disease ‐ Colony Rasmus Sejersten Ripa Stimulating Factor Therapy to Induce Neovascularization in Ischemic Heart Cover: Phoenix depicted in the book Disease Bilderbuch für Kinder by F.J. Bertuch (1747‐1822) ISBN 978‐87‐994776‐0‐9 FACULTY OF HEALTH SCIENCES UNIVERSITY OF COPENHAGEN Granulocyte-Colony Stimulating Factor Therapy to Induce Neovascularization in Ischemic Heart Disease Rasmus Sejersten Ripa Forsvaret finder sted onsdag den 30. november 2011, kl. 14 præcis i Medicinsk Museion, Bredgade 62, København Forsvarsleder: Professor, overlæge, dr.med. Torben V. Schroeder Formand for bedømmelsesudvalget: Professor, overlæge, dr.med. Niels Borregaard Universitetets officielle opponenter: Professor, overlæge, dr.med. Erling Falk og Professor, overlæge, ph.d., dr.med. Hans Erik Bøtker Denne afhandling er i forbindelse med anførte tidligere offentliggjorte artikler af Det Sundhedsvidenskabelige Fakultet ved Københavns Universitet antaget til offentligt at forsvares for den medicinske doktorgrad. København, den 1. august 2011. Ulla Wewer, Dekan This dissertation is based on the following original publications. These publications are referred to by their roman numerals: (I) Ripa RS, Wang Y, Jørgensen E, Johnsen HE, Hesse B, Kastrup J. Intramyocardial Injection of Vascular Endothelial Growth Factor-A165 Plasmid Followed by Granulocyte-Colony Stimulating Factor to Induce Angiogenesis in Patients with Severe Chronic Ischemic Heart Disease. Eur Heart J 2006;27:1785-92. (II) Ripa RS, Wang Y, Goetze JP, Jørgensen E, Johnsen HE, Tägil K, Hesse B, Kastrup J. Circulating Angiogenic Cytokines and Stem Cells in Patients with Severe Chronic Ischemic Heart Disease – Indicators of Myocardial Ischemic Burden? Int J Cardiol 2007;120:181-187. -

Advances in Biochemical Engineering/Biotechnology
SOFTbank E-Book Center Tehran, Phone: 66403879,66493070 For Educational Use. www.ebookcenter.ir 114 Advances in Biochemical Engineering/Biotechnology Series Editor: T. Scheper Editorial Board: W. Babel · I. Endo · S.-O. Enfors · M. Hoare ·W.-S. Hu B.Mattiasson · J. Nielsen · G. Stephanopoulos U. von Stockar · G. T. Tsao · R. Ulber · J.-J. Zhong SOFTbank E-Book Center Tehran, Phone: 66403879,66493070 For Educational Use. www.ebookcenter.ir Advances in Biochemical Engineering/Biotechnology Series Editor: T. Scheper Recently Published and Forthcoming Volumes Engineering of Stem Cells Tissue Engineering II Volume Editor: Martin, U. Basics of Tissue Engineering and Tissue Vol. 114, 2009 Applications Volume Editors: Lee, K., Kaplan, D. Biotechnology in China I Vol. 103, 2007 From Bioreaction to Bioseparation and Bioremediation Tissue Engineering I Volume Editors: Zhong, J.J., Bai, F.-W., Scaffold Systems for Tissue Engineering Zhang, W. Volume Editors: Lee, K., Kaplan, D. Vol. 113, 2009 Vol. 102, 2006 Bioreactor Systems for Tissue Engineering Cell Culture Engineering Volume Editors: Kasper, C., van Griensven, M., Volume Editor: Hu,W.-S. Poertner, R. Vol. 101, 2006 Vol. 112, 2008 Biotechnology for the Future Food Biotechnology Volume Editor: Nielsen, J. Volume Editors: Stahl, U., Donalies, U. E. B., Vol. 100, 2005 Nevoigt, E. Vol. 111, 2008 Gene Therapy and Gene Delivery Systems Protein – Protein Interaction Volume Editors: Schaffer, D.V., Zhou,W. Volume Editors: Seitz, H., Werther, M. Vol. 99, 2005 Vol. 110, 2008 Sterile Filtration Biosensing for the 21st Century Volume Editor: Jornitz, M.W. Volume Editors: Renneberg, R., Lisdat, F. Vol. 98, 2006 Vol. 109, 2007 Marine Biotechnology II Biofuels Volume Editors: Le Gal, Y., Ulber, R. -

Towards Mechanism- Based Treatments for Fragile X Syndrome
brain sciences Towards Mechanism- based Treatments for Fragile X Syndrome Edited by Daman Kumari and Inbal Gazy Printed Edition of the Special Issue Published in Brain Sciences www.mdpi.com/journal/brainsci Towards Mechanism-based Treatments for Fragile X Syndrome Towards Mechanism-based Treatments for Fragile X Syndrome Special Issue Editors Daman Kumari Inbal Gazy MDPI • Basel • Beijing • Wuhan • Barcelona • Belgrade Special Issue Editors Daman Kumari Inbal Gazy National Institute of Diabetes National Institute of Diabetes USA USA Editorial Office MDPI St. Alban-Anlage 66 4052 Basel, Switzerland This is a reprint of articles from the Special Issue published online in the open access journal Brain Sciences (ISSN 2076-3425) from 2018 to 2019 (available at: https://www.mdpi.com/journal/ brainsci/special issues/Fragile X Syndrome) For citation purposes, cite each article independently as indicated on the article page online and as indicated below: LastName, A.A.; LastName, B.B.; LastName, C.C. Article Title. Journal Name Year, Article Number, Page Range. ISBN 978-3-03921-505-8 (Pbk) ISBN 978-3-03921-506-5 (PDF) Cover image courtesy of Elisa A. Waxman, Children’s Hospital of Philadelphia, Philadelphia, PA, USA. c 2019 by the authors. Articles in this book are Open Access and distributed under the Creative Commons Attribution (CC BY) license, which allows users to download, copy and build upon published articles, as long as the author and publisher are properly credited, which ensures maximum dissemination and a wider impact of our publications. The book as a whole is distributed by MDPI under the terms and conditions of the Creative Commons license CC BY-NC-ND. -

DNA Methylation and Chromatin Dynamics During Postnatal Cardiac Development and Maturation Using in Vitro and in Vivo Model Systems
DNA methylation and chromatin dynamics during postnatal cardiomyocyte maturation Ms Sim Choon Boon B.Eng, M.Sc (Genetics) A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2017 School of Biomedical Sciences. Abstract Background: The neonatal mammalian heart has a transient capacity for regeneration, which is lost shortly after birth. A series of critical developmental transitions including a switch from hyperplastic to hypertrophic growth occur during this postnatal regenerative window, preparing the heart for the increased contractile demands of postnatal life. Postnatal cardiomyocyte maturation and loss of regenerative capacity are associated with expression alterations of thousands of genes embedded within tightly controlled transcriptional networks, which remain poorly understood. Interestingly, although mitogenic stimulation of neonatal cardiomyocytes results in proliferation, the same stimuli induce hypertrophy in adult cardiomyocytes by activating different transcriptional pathways, indicating that cardiomyocyte maturation may result from epigenetic modifications during development. Notably, DNA methylation and chromatin compaction are both important epigenetic modifications associated with a decrease in transcription factor (TF) accessibility to DNA. However, the role of both DNA methylation and chromatin compaction during postnatal cardiac maturation remain largely unknown. Hypothesis: Postnatal changes in DNA methylation and chromatin compaction silence transcriptional networks required