Towards Mechanism- Based Treatments for Fragile X Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Abstract Book – Oral Sessions
Sunday, June 21, 2015 12:00 p.m. - 1:15 p.m. Latin American Psychopharmacology Update: INNOVATIVE TREATMENTS IN PSYCHIATRY INNOVATIVE TREATMENTS IN PSYCHIATRY Flavio Kapczinski, The University of Texas Health Science Center at Houston Overall Abstract In this panel, we will propose innovative treatments in psychiatry and recent literature findings will be summarized. The effective pharmacological treatment of psychiatric diseases and development of new therapeutic entities has been a long-standing challenge. Despite the complexity and heterogeneity of psychiatric disorders, basic and clinical research studies and technological advancements in genomics, biomarkers, and imaging have begun to elucidate the pathophysiology of etiological complexity of psychiatric diseases and to identify efficacious new agents (Tcheremissine et al. 2014). Many psychiatric illnesses are associated with neuronal atrophy, characterized by loss of synaptic connections, increase of inflammatory markers like TNF-α and DAMPS (damage-associate molecular patterns,- cell free (ccf) DNA, heat shock proteins HSP70, HSP90, and HSP60, and cytochrome C), decrease of neurotrophic factors, increase of oxidative stress, mitochondrial dysfunction and apoptosis (Fries et al., 2012; Pfaffenseller et al., 2014). Also, neurocognitive impairment and poor psychosocial functioning has been related to a psychiatric disease. Therefore, trying to discover new therapeutic targets that act in these pathways can help to develop new treatment or improve the treatment of these serious diseases. -

Childhood Cerebral X-Linked Adrenoleukodystrophy with Atypical Neuroimaging Abnormalities and a No…
9/28/2018 Journal of Postgraduate Medicine: Childhood cerebral X-linked adrenoleukodystrophy with atypical neuroimaging abnormalities and a no… Open access journal indexed with Index Medicus & EMBASE Home | Subscribe | Feedback [Download PDF] CASE REPORT Year : 2018 | Volume : 64 | Issue : 1 | Page : 59-63 Childhood cerebral X-linked adrenoleukodystrophy with atypical neuroimaging abnormalities and a novel mutation M Muranjan1, S Karande1, S Sankhe2, S Eichler3, 1 Department of Pediatrics, Seth GS Medical College and KEM Hospital, Parel, Mumbai, Maharashtra, India 2 Department of Radiology, Seth GS Medical College and KEM Hospital, Parel, Mumbai, Maharashtra, India 3 Centogene AG, Schillingallee 68, Rostock, Germany Correspondence Address: Dr. M Muranjan Department of Pediatrics, Seth GS Medical College and KEM Hospital, Parel, Mumbai, Maharashtra India Abstract Childhood cerebral X-linked adrenoleukodystrophy (XALD) typically manifests with symptoms of adrenocortical insufficiency and a variety of neurocognitive and behavioral abnormalities. A major diagnostic clue is the characteristic neuroinflammatory parieto-occipital white matter lesions on magnetic resonance imaging. This study reports a 5-year 10-month old boy presenting with generalized skin hyperpigmentation since 3 years of age. Over the past 9 months, he had developed right-sided hemiparesis and speech and behavioral abnormalities, which had progressed over 5 months to bilateral hemiparesis. Retrospective analyses of serial brain magnetic resonance images revealed an unusual pattern of lesions involving the internal capsules, corticospinal tracts in the midbrain and brainstem, and cerebellar white matter. The clinical diagnosis of childhood cerebral adrenoleukodystrophy was confirmed by elevated basal levels of adrenocorticotropin hormone and plasma very long chain fatty acid levels. Additionally, sequencing of the ABCD1 gene revealed a novel mutation. -

The Effects and Feasibility of Using Tiered Instruction to Increase Conversational Turn Taking for Preschoolers with and Without Disabilities
THE EFFECTS AND FEASIBILITY OF USING TIERED INSTRUCTION TO INCREASE CONVERSATIONAL TURN TAKING FOR PRESCHOOLERS WITH AND WITHOUT DISABILITIES A dissertation submitted to the Kent State University College and Graduate School of Education, Health, and Human Services in partial fulfillment of the requirements for the degree of Doctor of Philosophy By Sandra Hess Robbins May 2013 © Copyright, 2013 by Sandra Hess Robbins All Rights Reserved ii A dissertation written by Sandra Hess Robbins B.S.Ed., University of Wisconsin, Oshkosh, 2002 M.Ed., Kent State University, 2006 Ph.D., Kent State University, 2013 Approved by ____________________, Co-director, Doctoral Dissertation Committee Kristie Pretti-Frontcak ____________________, Co-director, Doctoral Dissertation Committee Sanna Harjusola-Webb ____________________, Member, Doctoral Dissertation Committee Christine Balan ____________________, Member, Doctoral Dissertation Committee Richard Cowan Accepted by ____________________, Director, School of Lifespan Development and Educational Mary Dellman-Jenkins Sciences ____________________, Dean, College of Education, Health, and Human Services Daniel F. Mahony iii ROBBINS, SANDRA HESS, Ph.D., May 2013 Lifespan Development and Educational Sciences THE EFFECTS AND FEASIBILITY OF USING TIERED INSTRUCTION TO INCREASE CONVERSATIONAL TURN TAKING FOR PRESCHOOLERS WITH AND WITHOUT DISABILITIES (282 pp.) Co-Directors of Dissertation: Kristie Pretti-Frontczak, Ph.D. Sanna Harjusola-Webb, Ph.D. The purpose of the study was to examine the effectiveness and feasibility of using tiered instruction to increase the frequency of conversational turn taking (CTT) among preschoolers with and without disabilities in an inclusive setting. Three CTT interventions (Universal Design for Learning, Peer Mediated Instruction, and Milieu Teaching) were organized on a hierarchy of intensity and implemented in an additive manner. -

Ryerson University Spring Graduates
Ryerson University Spring Graduates June 2020 Faculty of Arts 2 Faculty of Communication & Design 11 Faculty of Community Services 21 Faculty of Engineering and Architectural Science 35 Faculty of Science 46 Ted Rogers School of Management 54 Yeates School of Graduate Studies 71 The G. Raymond Chang School of Continuing Education 73 Faculty of Arts Pamela Sugiman Dean Faculty of Arts Janice Fukakusa Chancellor Mohamed Lachemi President and Vice-Chancellor Charmaine Hack Registrar Ryerson Gold Medal Presented to Mayah Obadia Geographic Analysis 2 Faculty of Arts Undergraduate Degree Programs Arts and Contemporary Studies Bachelor of Arts (Honours) *Diana Abo Harmouch Carmen Jajjo *Megumi Noteboom *Sima Rebecca Abrams Leya Jasat Valentina Padure Qeyam Amiri Sophie Johnson *Naiomi Marcia Perera Brodie Barrick Babina Kamalanathan Charlotte Jane Prokopec Rebecca Claire Chen Caroline Susan Kewley Regan Reynolds Erin Tanya Clarke Jessica Laurenza Joshua Ricci *Megan Lisa Devoe Claire Lowenstein Kaitlin Anganie Seepersaud *Manpreet Kaur Dhaliwal *Avigayil Margolis Gabriela Skwarko Tatum Lynn Donovan Sara McArthur Julia Macey Sullivan Faith Raha Giahi *Nadia Celeste McNairn *Helen Gillian Webb Meagan Gove *Mahbod Mehrvarz *Michael Worbanski Salem Habtom Andrew Moon Smyrna Wright *William Hanchar *Liana Gabriella Mortin Calum Jacques Potoula Mozas Criminology Bachelor of Arts (Honours) *Annabelle Adjei *Jenna Anne Giannini Veronica Hiu Lam Lee Stanislav Babinets Albina Glatman Karishma Catherine Lutchman Hela Bakhtari Farah Khaled Gregni Simbiat -

Nicole Trask, Pharmd, Planning for the 2019 Specialty Drug Spend
Planning for the 2019 Specialty Drug Spend August 24, 2018 Nicole Trask, PharmD Clinical Consultant Pharmacist University of Massachusetts – Clinical Pharmacy Services Disclosure for Nicole Trask I have no actual or potential conflict of interest in relation to this presentation. Budget Impact Modeling for 2 ||August 24, 2018 the Specialty Drug Spend Objectives • Identify high-impact specialty pipeline drugs expected to reach the market in 2019-2020 • Summarize efficacy data for high-impact specialty pipeline drugs and indicate their anticipated place in therapy • Compare specialty pipeline drugs to currently available therapeutic options • Predict the budgetary impact of specialty pipeline drugs and discuss strategies to mitigate costs Budget Impact Modeling for 3 ||August 24, 2018 the Specialty Drug Spend Identifying High-Impact Drugs Two key drivers • Clinical impact • Efficacy/effectiveness • Therapeutic alternatives • Economic impact • Cost • Volume Budget Impact Modeling for 4 ||August 24, 2018 the Specialty Drug Spend Assessing Clinical Impact Clinical trial data Therapeutic alternatives • Placebo-controlled, • Me-too drug vs. head-to-head studies first-in-class • Adverse events • Market competition • Potential drug-drug • Consensus interactions guidelines • Target population • Patient willingness to use medication Budget Impact Modeling for 5 ||August 24, 2018 the Specialty Drug Spend Assessing Economic Impact Cost Volume • NADAC, AWP, WAC • Prevalence/incidence of • Supplemental rebate disease • Outcomes-based • Frequency of contracts administration • Value assessments • Duration of therapy (e.g., AHRQ, ICER, PCORI) AHRQ=Agency for Healthcare Research and Quality, AWP=average wholesale price, ICER=Institute for Clinical and Economic Review, NADAC=national average drug acquisition cost, PCORI=Patient-centered Outcomes Research Institute, WAC=wholesale acquisition cost Budget Impact Modeling for 6 ||August 24, 2018 the Specialty Drug Spend Other Factors Affecting Budget Impact Disease-specific Prescriber-specific • Chronic vs. -

Women with High Functioning ASD: Relationships and Sexual Health
Claremont Colleges Scholarship @ Claremont CMC Senior Theses CMC Student Scholarship 2020 Women with High Functioning ASD: Relationships and Sexual Health Isabelle Taylor Follow this and additional works at: https://scholarship.claremont.edu/cmc_theses Part of the Developmental Psychology Commons Recommended Citation Taylor, Isabelle, "Women with High Functioning ASD: Relationships and Sexual Health" (2020). CMC Senior Theses. 2553. https://scholarship.claremont.edu/cmc_theses/2553 This Open Access Senior Thesis is brought to you by Scholarship@Claremont. It has been accepted for inclusion in this collection by an authorized administrator. For more information, please contact [email protected]. Claremont McKenna College Women with High Functioning ASD: Relationships and Sexual Health A Literature Review and Proposed Workshop Manual Submitted to Professor Caitlyn Gumaer By Isabelle Taylor for Senior Thesis 2020 November 30 Table of Contents Author’s Note.................................................................................................................................. 1 Abstract........................................................................................................................................... 2 Literature Review.................................................................................................................... 3 - 38 Symptoms and Diagnosis.............................................................................................. 3 - 7 Gender Differences..................................................................................................... -

Rett Syndrome: Coming to Terms with Treatment
Hindawi Publishing Corporation Advances in Neuroscience Volume 2014, Article ID 345270, 20 pages http://dx.doi.org/10.1155/2014/345270 Review Article Rett Syndrome: Coming to Terms with Treatment Alan Percy Civitan International Research Center, University of Alabama at Birmingham, 1720 2nd Avenue South, CIRC 320E, Birmingham, AL 35294-0021, USA Correspondence should be addressed to Alan Percy; [email protected] Received 5 January 2014; Accepted 26 February 2014; Published 10 April 2014 Academic Editor: Ronald L. Klein Copyright © 2014 Alan Percy. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Rett syndrome (RTT) has experienced remarkable progress over the past three decades since emerging as a disorder of worldwide proportions, particularly with discovery of the linkage of RTT to MECP2 mutations. The advances in clinical research and the increasing pace of basic science investigations have accelerated the pattern of discovery and understanding. Clinical trials are ongoing and others are planned. A review of these events and the prospects for continued success are highlighted below. The girls and women encountered today with RTT are, overall, in better general, neurologic, and behavioral health than those encountered earlier. This represents important progress worldwide from the concerted efforts of a broadly based and diverse clinical and basic research consortium as well -

FY19 Annual Report View Report
Annual Report 2018–19 3 Introduction 5 Metropolitan Opera Board of Directors 6 Season Repertory and Events 14 Artist Roster 16 The Financial Results 20 Our Patrons On the cover: Yannick Nézet-Séguin takes a bow after his first official performance as Jeanette Lerman-Neubauer Music Director PHOTO: JONATHAN TICHLER / MET OPERA 2 Introduction The 2018–19 season was a historic one for the Metropolitan Opera. Not only did the company present more than 200 exiting performances, but we also welcomed Yannick Nézet-Séguin as the Met’s new Jeanette Lerman- Neubauer Music Director. Maestro Nézet-Séguin is only the third conductor to hold the title of Music Director since the company’s founding in 1883. I am also happy to report that the 2018–19 season marked the fifth year running in which the company’s finances were balanced or very nearly so, as we recorded a very small deficit of less than 1% of expenses. The season opened with the premiere of a new staging of Saint-Saëns’s epic Samson et Dalila and also included three other new productions, as well as three exhilarating full cycles of Wagner’s Ring and a full slate of 18 revivals. The Live in HD series of cinema transmissions brought opera to audiences around the world for the 13th season, with ten broadcasts reaching more than two million people. Combined earned revenue for the Met (box office, media, and presentations) totaled $121 million. As in past seasons, total paid attendance for the season in the opera house was 75%. The new productions in the 2018–19 season were the work of three distinguished directors, two having had previous successes at the Met and one making his company debut. -

Pelizaeus-Merzbacher Disease (Pmd)
PELIZAEUS‐MERZBACHER DISEASE (PMD) is a X‐linked disease that is transmitted from normal appearing carrier mothers to their sons. Each male born to these mothers has a 50/50 chance of being affected with PMD. Each female child born to them has a 50/50 chance of being a carrier herself. Having more than one affected child, with a disease that there is only 50/50 of, I know sounds impossible, but I did the same thing! You have to realize it's a 50/50 chance with every pregnancy. One way to possibly describe this is to take a deck of cards, remove all 4 aces. Let the 2 red ones represent females, the ace of hearts could represent a non‐carrier female, the ace of diamonds could represent a carrier female. Let the 2 black ones represent males, the ace of clubs could represent a non‐affected male, the ace of spades could represent an affected male. With each pregnancy you have a 1 in 4 chance of having an affected son. Turn the 4 cards face down and draw one, if it should be the ace of spades (affected male), you can't say, "I have already had my affected one", and exclude it from the next pregnancy. With each pregnancy you have to "draw" from all 4 "cards". This disease affects the myelin sheath, that insulates the nerves of the central nervous system. These nerve fibers, called axons, carry impulses from the brain to other parts of the body. The axons are similar to an electrical wire, the myelin is like insulation that covers them. -

X-Linked Diseases: Susceptible Females
REVIEW ARTICLE X-linked diseases: susceptible females Barbara R. Migeon, MD 1 The role of X-inactivation is often ignored as a prime cause of sex data include reasons why women are often protected from the differences in disease. Yet, the way males and females express their deleterious variants carried on their X chromosome, and the factors X-linked genes has a major role in the dissimilar phenotypes that that render women susceptible in some instances. underlie many rare and common disorders, such as intellectual deficiency, epilepsy, congenital abnormalities, and diseases of the Genetics in Medicine (2020) 22:1156–1174; https://doi.org/10.1038/s41436- heart, blood, skin, muscle, and bones. Summarized here are many 020-0779-4 examples of the different presentations in males and females. Other INTRODUCTION SEX DIFFERENCES ARE DUE TO X-INACTIVATION Sex differences in human disease are usually attributed to The sex differences in the effect of X-linked pathologic variants sex specific life experiences, and sex hormones that is due to our method of X chromosome dosage compensation, influence the function of susceptible genes throughout the called X-inactivation;9 humans and most placental mammals – genome.1 5 Such factors do account for some dissimilarities. compensate for the sex difference in number of X chromosomes However, a major cause of sex-determined expression of (that is, XX females versus XY males) by transcribing only one disease has to do with differences in how males and females of the two female X chromosomes. X-inactivation silences all X transcribe their gene-rich human X chromosomes, which is chromosomes but one; therefore, both males and females have a often underappreciated as a cause of sex differences in single active X.10,11 disease.6 Males are the usual ones affected by X-linked For 46 XY males, that X is the only one they have; it always pathogenic variants.6 Females are biologically superior; a comes from their mother, as fathers contribute their Y female usually has no disease, or much less severe disease chromosome. -

Brain Metabolic Profile After Intranasal Vs. Intraperitoneal Clomipramine
International Journal of Molecular Sciences Article Brain Metabolic Profile after Intranasal vs. Intraperitoneal Clomipramine Treatment in Rats with Ultrasound Model of Depression Olga Abramova 1,2, Yana Zorkina 1,2,* , Timur Syunyakov 2,3 , Eugene Zubkov 1, Valeria Ushakova 1,2, Artemiy Silantyev 1, Kristina Soloveva 2, Olga Gurina 1, Alexander Majouga 4, Anna Morozova 1,2 and Vladimir Chekhonin 1,5 1 V. Serbsky National Medical Research Centre of Psychiatry and Narcology, 119034 Moscow, Russia; [email protected] (O.A.); [email protected] (E.Z.); [email protected] (V.U.); [email protected] (A.S.); [email protected] (O.G.); [email protected] (A.M.); [email protected] (V.C.) 2 Mental-Health Clinic No. 1 Named after N.A. Alekseev, 117152 Moscow, Russia; [email protected] (T.S.); [email protected] (K.S.) 3 Federal State Budgetary Institution Research Zakusov Institute of Pharmacology, 125315 Moscow, Russia 4 Drug Delivery Systems Laboratory, D. Mendeleev University of Chemical Technology of Russia, Miusskaya pl. 9, 125047 Moscow, Russia; [email protected] 5 Department of Medical Nanobiotechnology, Pirogov Russian National Research Medical University, 117997 Moscow, Russia * Correspondence: [email protected]; Tel.: +7-916-588-4851 Citation: Abramova, O.; Zorkina, Y.; Abstract: Background: Molecular mechanisms of depression remain unclear. The brain metabolome Syunyakov, T.; Zubkov, E.; Ushakova, after antidepressant therapy is poorly understood and had not been performed for different routes V.; Silantyev, A.; Soloveva, K.; Gurina, of drug administration before the present study. Rats were exposed to chronic ultrasound stress O.; Majouga, A.; Morozova, A.; et al. and treated with intranasal and intraperitoneal clomipramine. -
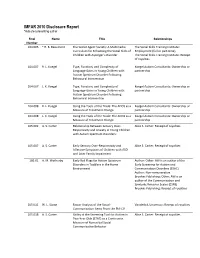
IMFAR 2010 Disclosure Report *Indicates Presenting Author
IMFAR 2010 Disclosure Report *indicates presenting author Final Name Title Relationships Number 104.005 * R. B. Beaumont The Secret Agent Society: A Multimedia The Social Skills Training Institute: Curriculum for Enhancing the Social Skills of Employment (full or part-time) Children with Asperger's Disorder The Social Skills Training Institute: Receipt of royalties 104.007 R. L. Koegel Type, Function, and Complexity of Koegel Autism Consultants: Ownership or Language Gains in Young Children with partnership Autism Spectrum Disorder Following Behavioral Intervention 104.007 L. K. Koegel Type, Function, and Complexity of Koegel Autism Consultants: Ownership or Language Gains in Young Children with partnership Autism Spectrum Disorder Following Behavioral Intervention 104.008 R. L. Koegel Using the Tools of the Trade: The ADOS as a Koegel Autism Consultants: Ownership or Measure of Treatment Change partnership 104.008 L. K. Koegel Using the Tools of the Trade: The ADOS as a Koegel Autism Consultants: Ownership or Measure of Treatment Change partnership 105.002 A. S. Carter Relationship Between Sensory Over- Alice S. Carter: Receipt of royalties Responsivity and Anxiety in Young Children with Autism Spectrum Disorders: 105.007 A. S. Carter Early Sensory Over-Responsivity and Alice S. Carter: Receipt of royalties Affective Symptoms of Children with ASD and Later Family Impairment 105.01 A. M. Wetherby Early Red Flags for Autism Spectrum Author: Other: AW is an author of the Disorders in Toddlers in the Home Early Screening for Autism and Environment Communication Disorders (ESAC) Author: Non-remunerative Brookes Publishing: Other: AW is an author of the Communication and Symbolic Behavior Scales (CSBS) Brookes Publishing: Receipt of royalties 105.011 W.