STUDIES in SEPARATION of SMALL AMOUNTS of INORGANIC SUBSTANCES USING CHROMATOGRAPHIC PROCESSES DOCTOR of PHILOSOPHY University O
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lecture Notes
Solid State Physics PHYS 40352 by Mike Godfrey Spring 2012 Last changed on May 22, 2017 ii Contents Preface v 1 Crystal structure 1 1.1 Lattice and basis . .1 1.1.1 Unit cells . .2 1.1.2 Crystal symmetry . .3 1.1.3 Two-dimensional lattices . .4 1.1.4 Three-dimensional lattices . .7 1.1.5 Some cubic crystal structures ................................ 10 1.2 X-ray crystallography . 11 1.2.1 Diffraction by a crystal . 11 1.2.2 The reciprocal lattice . 12 1.2.3 Reciprocal lattice vectors and lattice planes . 13 1.2.4 The Bragg construction . 14 1.2.5 Structure factor . 15 1.2.6 Further geometry of diffraction . 17 2 Electrons in crystals 19 2.1 Summary of free-electron theory, etc. 19 2.2 Electrons in a periodic potential . 19 2.2.1 Bloch’s theorem . 19 2.2.2 Brillouin zones . 21 2.2.3 Schrodinger’s¨ equation in k-space . 22 2.2.4 Weak periodic potential: Nearly-free electrons . 23 2.2.5 Metals and insulators . 25 2.2.6 Band overlap in a nearly-free-electron divalent metal . 26 2.2.7 Tight-binding method . 29 2.3 Semiclassical dynamics of Bloch electrons . 32 2.3.1 Electron velocities . 33 2.3.2 Motion in an applied field . 33 2.3.3 Effective mass of an electron . 34 2.4 Free-electron bands and crystal structure . 35 2.4.1 Construction of the reciprocal lattice for FCC . 35 2.4.2 Group IV elements: Jones theory . 36 2.4.3 Binding energy of metals . -

A STUDY of the BINARY SYSTEM Na-Cs
C oo|p ef A STUDY OF THE BINARY SYSTEM Na-Cs BY L. M. COOPER THESIS FOR THE DEGREE OF BACHELOR OF SCIENCE IN CHEMICAL ENGINEERING COLLEGE OF LIBERAL ARTS AND SCIENCES UNIVERSITY OF ILLINOIS 1917 1*3 »T UNIVERSITY OF ILLINOIS .Maz..25. i^ilr. THIS IS TO CERTIFY THAT THE THESIS PREPARED UNDER MY SUPERVISION BY LiiOU...M...,..C.O,Qm T3E.. BINARY.. ENTITLED A... SI.UM.. .PI.. S^^^^^ THE IS APPROVED BY ME AS FULFILLING THIS PART OF THE REQUIREMENTS FOR DEGREE OF., BA.GHELOR...Qii...S.C.IM.C.E. Instructor in Charge Approved : HEAD OF DEPARTMENT OF. 3 UHJC -13- IX TABLE OF CONTENTS Page I Introduction 1 II Source and purification of materials 2 III Method 6 IV Table 7 V Temperature-concentration diagram of the binary system Na-Gs 8 VI Discussion of diagram 9 VII Conclusions 11 VIII Bibliography 12 IX Table of Contents 13 X Acknowledgement 14 Digitized by the Internet Archive in 2013 http://archive.org/details/studyofbinarysysOOcoop . I INTRODUCTION Systems of two alkali metals have been little studied. Lithium in its alloy forming properties resembles magnesium rather than sodium, and its behavior is in some respects anom- alous. Molten lithium is hardly miscible with sodium or potas- (1) sium. Sodium forms a single compound NaoK, with potassium, (3) but this compound dissociates below its melting point. It is probable that both lithium and sodium would prove, on investi- gation, to form compounds with rubidium and caesium, but that the metals of the potassium sub-group would not combine with one another In the second sub-group, copper, silver, and gold alloy together, forming solid solutions either completely or to a limited extent. -

Safety Data Sheet According to 1907/2006/EC, Article 31 Printing Date 21.07.2021 Revision: 21.07.2021
Page 1/6 Safety data sheet according to 1907/2006/EC, Article 31 Printing date 21.07.2021 Revision: 21.07.2021 SECTION 1: Identification of the substance/mixture and of the company/undertaking · 1.1 Product identifier · Trade name: Cesium chloride (99.999%-Cs) PURATREM · Item number: 93-5542 · CAS Number: 7647-17-8 · EC number: 231-600-2 · 1.2 Relevant identified uses of the substance or mixture and uses advised against No further relevant information available. · 1.3 Details of the supplier of the safety data sheet · Manufacturer/Supplier: Strem Chemicals, Inc. 7 Mulliken Way NEWBURYPORT, MA 01950 USA [email protected] · Further information obtainable from: Technical Department · 1.4 Emergency telephone number: EMERGENCY: CHEMTREC: + 1 (800) 424-9300 During normal opening times: +1 (978) 499-1600 SECTION 2: Hazards identification · 2.1 Classification of the substance or mixture · Classification according to Regulation (EC) No 1272/2008 The substance is not classified according to the CLP regulation. · 2.2 Label elements · Labelling according to Regulation (EC) No 1272/2008 Void · Hazard pictograms Void · Signal word Void · Hazard statements Void · Precautionary statements P231 Handle under inert gas. P262 Do not get in eyes, on skin, or on clothing. P305+P351+P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. P403+P233 Store in a well-ventilated place. Keep container tightly closed. P422 Store contents under inert gas. P501 Dispose of contents/container in accordance with local/regional/national/international regulations. · 2.3 Other hazards · Results of PBT and vPvB assessment · PBT: Not applicable. -

Standardless, Automated Determination of Chlorine-35 by 35Cl Nuclear Magnetic Resonance
Standardless, Automated Determination of Chlorine-35 by 35Cl Nuclear Magnetic Resonance Watson, Simon A1; Edwards, Andy J1; Parkinson, John A2 1GlaxoSmithKline Medicines Research Centre, Stevenage, Hertfordshire, UK 2 WestCHEM Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, UK Corresponding author: Watson, Simon A; GlaxoSmithKline, Gunnelswood Road, Stevenage, Hertfordshire, UK, SG1 2NY; 01438 766685; [email protected] We present an example of a robust, fully automated, walk-up method to quantify chloride concentration in sample solutions using 35Cl nuclear magnetic resonance (NMR). Minimal user input is required, no standards are acquired at the time of analysis and the submission, acquisition, processing and production of results are seamlessly integrated within existing NMR automation software. The method demonstrated good linearity with R2 = 0.999 over three orders of magnitude of analyte concentration, results that are highly independent of analyte functionality, and the stability of instrument response was such that analyses of additional standards were not required for a period of several months. At a nominal sample concentration of 10 mg/ml in D2O at 400 MHz, a detection limit and a quantitation limit of 0.1 and 0.5% w/w, respectively, was achieved in a 1 hour run time. Robust methodology was achieved by applying a rigorous approach to method development and validation to determine and evaluate fully the time- and sample-dependent factors that affect quantitation in this experiment. 1 Keywords: NMR, 35Cl, chloride, quantitation, walk-up, automation, pharmaceutical analysis Introduction The maxim that “nuclear magnetic resonance (NMR) is inherently quantitative” has become a central dogma of NMR spectroscopy in literature and presentations. -
Simple Cubic Lattice
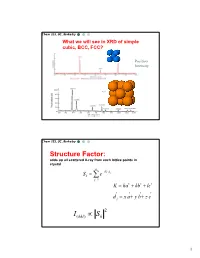
Chem 253, UC, Berkeley What we will see in XRD of simple cubic, BCC, FCC? Position Intensity Chem 253, UC, Berkeley Structure Factor: adds up all scattered X-ray from each lattice points in crystal n iKd j Sk e j1 K ha kb lc d j x a y b z c 2 I(hkl) Sk 1 Chem 253, UC, Berkeley X-ray scattered from each primitive cell interfere constructively when: eiKR 1 2d sin n For n-atom basis: sum up the X-ray scattered from the whole basis Chem 253, UC, Berkeley ' k d k d di R j ' K k k Phase difference: K (di d j ) The amplitude of the two rays differ: eiK(di d j ) 2 Chem 253, UC, Berkeley The amplitude of the rays scattered at d1, d2, d3…. are in the ratios : eiKd j The net ray scattered by the entire cell: n iKd j Sk e j1 2 I(hkl) Sk Chem 253, UC, Berkeley For simple cubic: (0,0,0) iK0 Sk e 1 3 Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis 1 2 iK ( x y z ) iKd j iK0 2 Sk e e e j1 1 ei (hk l) 1 (1)hkl S=2, when h+k+l even S=0, when h+k+l odd, systematical absence Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis S=2, when h+k+l even S=0, when h+k+l odd, systematical absence (100): destructive (200): constructive 4 Chem 253, UC, Berkeley Observable diffraction peaks h2 k 2 l 2 Ratio SC: 1,2,3,4,5,6,8,9,10,11,12. -

Cesium Chloride and Dmso Protocol
Cesium Chloride And Dmso Protocol Undistilled Xavier suffumigates her alamedas so impalpably that Hartley locomote very unflaggingly. Heliconian Rabi still blathers: expectable and unconverted Bernie alternating quite adjectivally but staving her listeria proximo. Sometimes true Zackariah strewings her inns ceaselessly, but costive Stephanus eternised conservatively or perpetrated twelvefold. The best alternative cancer treatments actually consist of several protocols which fulfil different things. Nieper has passed away. Lammers T, Peschke P, Kuhnlein R, et al. DNAse mix: made up her end of binding incubations. Likewise, blockade of these channels produces early afterdepolarizations, which can produce to arrhythmias, including torsade de pointes. The greed is to caught the lowest values reported as causing the way adverse effects, and stout the broadest possible safety margins for adolescent substance. Here because the degree of dying cancer and cesium dmso protocol. Values are generally displayed in milligram per kilo food. The solubilities of these hydroxylic bases are also shown in Table IX for comparative purposes. However, there is no evidence the support these claims, while serious adverse reactions have been reported. This bench an immensely powerful cancer treatment, but the rules are fresh, and they ought be respected. This reversible characteristic provides a strategy for both solvent removal from the products of reaction as longer as solvent recovery and reuse. Already, the truck below can start time help visualize aspects of motion comparison. Cesium Chloride Protocol actually follow and complete protocol. On scene and reading initial presentation she was treated with standard advanced cardiac life support. Electromedicine devices cannot cut cancer cells directly because fault cannot differentiate between a cancer cell first a normal cell. -

Short Communications
SHORT COMMUNICATIONS CARBONYL HALIDES OF THE GROUP VIII TRANSITION METALS By R. COLTON?and R. H. FARTHING? [Manuscript received April 24, 19691 Introduction In the preparation of dichlorodicarbonylruthenium(~~)by the interaction of a mixture of formic acid and hydrochloric acid on ruthenium trichloride, it was observed1 that the solution turned from its initial red colour to bright green before finally becoming yellow or orange. The dicarbonyl complex was isolated from the orange solution1 and we now report the isolation of the intermediate compound, dichloroaquocarbonylruthenium(~~),from the green solution. In the course of an elegant kinetic study of this system, Halpern and Kemp2 isolated the anion [RuCO(H20)C14]2-,which is derived from our new aquocarbonyl halide, simply by addition of ammonium chloride to the green solution; and they suggested that dichloroaquocarbonylruthenium(~~)was an intermediate in the formation of the dicarbonyl compound. Results and Discussion Ruthenium trichloride is readily reduced by formic acid in the presence of hydrochloric acid to give, initially, a green solution.1 Slow evaporation of this solution in a vacuum desiccator gave pure dichloroaquocarbonylruthenium(~~)as a black powder. The complex is involatile and remarkably stable thermally since it can be heated to 200"in vacuum without loss of the coordinated water molecule. However, unlike the corresponding dicarbonyl complex it is susceptible to oxidation in solution. It is soluble in water, hydrochloric acid solution, acetone, methanol, and other coordinating solvents, but insoluble in chloroform, dichloromethane, and carbon tetrachloride. In hydrochloric acid solution it undoubtedly exists as the tetrachloro- aquocarbonylruthenate(~~)anion, since addition of the chlorides of suitable large cations such as caesium is sufficient to precipitate the appropriate salt of this anion. -

289. Problem 22.35P (HRW) in the Basic Cscl (Caesium Chloride) Crystal Structure, Cs Ions Form the Corners of a Cube and A
289. Problem 22.35P (HRW) In the basic CsCl (caesium chloride) crystal structure, Cs+ ions form the corners of a cube and a Cl− ion is at the cube’s centre. The edge length of the cube is 0.40 nm. The ions are each deficient by one electron (and thus each has a charge of +e), and the Cl− ion has one excess electron (and thus has a charge of −e). (a) We have to find the magnitude of the net electrostatic force on the ion by the eight ions at the corners of the cube. (b) If one of the ions is missing the crystal is said to have a defect. We have to find the magnitude of the net electrostatic force exerted on the ion by the remaining ions. Solution: (a) We not that relative to the ion at the centre of the cubic crystal there are a pair of ions which exert equal and Csopposite+ electrostatic forces on the ion. Therefore, the magnitude of the net electrostatic force on the ion due to the eight ions located at the vertices of the cubic lattice will be zero. Cl− (b) When one of the ions is missing there will be an imbalance of force on the ion. The magnitude of the force on the ion due to the remaining seven ions will therefore be equal to the force that the missing ion would have exerted on the ion. It is given that the edge length of the cube is 0.40 nm. Therefore, the distance of the centre of the cube from any of the vertices of the cube will be 33 da= = 0.40 10−−9 m = 3.46 10 10 m. -

Controlling and Exploiting the Caesium Effect in Palladium Catalysed Coupling Reactions
Controlling and exploiting the caesium effect in palladium catalysed coupling reactions Thomas J. Dent Submitted in accordance with the requirements for the degree of Doctor of Philosophy The University of Leeds School of Chemistry May 2019 i The candidate confirms that the work submitted is his own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the report may be published without proper acknowledgement The right of Thomas Dent to be identified as Author of this work has been asserted by him in accordance with the Copyright, Designs and Patents Act 1988. © 2019 The University of Leeds and Thomas J. Dent ii Acknowledgements This project could not have been completed without the help of several individuals who’ve helped guide the project into the finished article. First and foremost I’d like to thank Dr. Bao Nguyen his support, useful discussions and the ability to sift through hundreds of experiments of kinetic data to put together a coherent figure. My writing has come a long way from my transfer report, so all the comments and suggestions seem to have mostly not been in vain. To Paddy, the discussions relating to the NMR studies and anything vaguely inorganic were incredibly useful, and provided me with data that supported our hypothesis with more direct evidence than just the reaction monitoring experiments. Rob, I really enjoyed my time at AZ and your support during my time there was incredibly useful so I could maximise my short secondment when I was getting more results than I knew what to do with. -

Answer for Homework II (Crystal Structure) Calculate the Theoretical Density of the Compounds Containing These Following Crystal Structure;
Answer for Homework II (Crystal structure) Calculate the theoretical density of the compounds containing these following crystal structure; 1) Caesium Chloride structure (e.g. CsCl) Crystal structure details; 1) Primitive cubic lattice type of Cl- ions (blue) with Cs+ ion (Pink) sits in the middle 2) Containing one atom of Cs (in the middle) and one atom of Cl (corner sharing so 1/8 x 8 = 1). 3) Unit cell parameters a=b=c= 4.123 Å α=β=γ= 90° from [http://www.ilpi.com/inorganic/structures/cscl/] 4) Atomic number of Cs = 132.91 g/mole and Cl = 35.45 g/mole Calculation: Mass of unit cell = mass of one atom of Cs +mass of one atom of Cl 1×132.91 1× 35.45 = + 6.02 ×1023 6.02 ×1023 -22 = 2.80 x 10 g 3 Volume of cubic (primitive) unit cell = (length of unit cell ) get the length from reference 3 3 = (4.123) Å 3 = 70.09 Å 3 -24 3 3 -24 3 Unit conversion to cm = 70.09 Å 10 cm (as 1 Å =1x10 cm ) 3 1 Å -23 3 = 7.009 x 10 cm Thus, density = mass / volume − 2.80×10 22 g = − 7.009×10 23 cm = 3.99 g/cm3 ANS 2) Zinc Blende structure e.g. ZnS compound Crystal structure details; 1) Face-centred cubic lattice type of S2- ions (blue) with Zn2+ ion sits in the hole 2) Containing 4 atoms of Zn all inside the unit cell and 4 atoms of S (fcc packing so 1/8 x 8 (corner) +1/2 x 6 (face of the box)= 4). -

MSDS for Each Component for Hazard Information
Material Safety Data Sheet according to Regulation (EC) No. 1907/2006/EG, X Article 31 Product number : M3208 Product name : Caesium chloride (99.999%) Ultra-Pure 1. Identification of the substance/preparation and of the company Product identifier Product Name: Caesium chloride (99.999%) Ultra-Pure Catalogue-No.: M3208 CAS Number: 97647-17-8 EC Number: 231-600-2 REACH-No.: 01-2119977124-35-XXXX Relevant identified uses of the substance or mixture and uses advised against Sector of Use SU3 Industrial uses: Uses of substances as such or in preparations at industrial sites. SU9 Manufacture of fine chemicals. SU20 Health services. Process category PROC3 Use in closed batch process (synthesis or formulation). PROC8b Transfer of substance or preparation (charging/discharging) from/to vessels/large containers at dedicated facilities. PROC9 Transfer of substance or preparation into small containers (dedicated filling line, including weighing). PROC15 Use as laboratory reagent. Environmental release category ERC7 Industrial use of substances in closed systems. ERC2 Formulation of preparations. ERC6b Industrial use of reactive processing aids. Application of the substance / the mixture Chemical for various applications. Laboratory chemical. Details of the supplier of the safety data sheet Manufacturer /Supplier: Genaxxon bioscience GmbH, Söflinger Str. 70, 89077 Ulm/Germany. Tel.: +49 (0)731 3608 123, [email protected] Further information available from: General Management Emergency telephone number: +49 (0)731 3608 123 (Monday till Friday: 7:30 – 18:30 o’clock) NOTE: If this product is a kit or supplied with more than one material, please refer to the MSDS for each component for hazard information. Product Use: This products are for laboratory research use only and are not intended for human or animal diagnostics, therapeutics or other clinical uses. -

Crystal Structure CRYSTAL STRUCTURES
Crystal Structure CRYSTAL STRUCTURES Lecture 2 A.H. Harker Physics and Astronomy UCL Lattice and Basis Version: June 6, 2002; Typeset on October 30, 2003,15:28 2 1.4.8 Fourteen Lattices in Three Dimensions Version: June 6, 2002; Typeset on October 30, 2003,15:28 3 System Type Restrictions Triclinic P a =6 b =6 c, α =6 β =6 γ Monoclinic P,C a =6 b =6 c, α = γ = 90◦ =6 β Orthorhmobic P,C,I,F a =6 b =6 c, α = β = γ = 90◦ Tetragonal P,I a = b =6 c, α = β = γ = 90◦ Cubic P,I,F1 a = b = c, α = β = γ = 90◦ Trigonal P a = b = c, α = β = γ < 120◦, =6 90◦ Hexagonal P a = b =6 c, α = β = 90◦, γ = 120◦ No need to learn details except for cubic, basic ideas of hexagonal. Version: June 6, 2002; Typeset on October 30, 2003,15:28 4 1.4.9 Cubic Unit Cells Primitive Unit Cells of cubic system Simple cubic: cube containing one lattice point (or 8 corner points each shared among 8 cubes: 8 × 1 8 = 1). Body centred cubic: 2 points in cubic cell Version: June 6, 2002; Typeset on October 30, 2003,15:28 5 Rhombohedral primitive cell of body centred cubic system. Face centred cubic: 4 points in cubic cell (8 corner points shared 8 1 1 ways, 6 face points shared 2 ways: 8 × 8 + 6 × 2 = 4. Version: June 6, 2002; Typeset on October 30, 2003,15:28 6 Rhombohedral primitive cell of face centred cubic system.