Crystal Physics and X Ray Diffraction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

361: Crystallography and Diffraction
361: Crystallography and Diffraction Michael Bedzyk Department of Materials Science and Engineering Northwestern University February 2, 2021 Contents 1 Catalog Description3 2 Course Outcomes3 3 361: Crystallography and Diffraction3 4 Symmetry of Crystals4 4.1 Types of Symmetry.......................... 4 4.2 Projections of Symmetry Elements and Point Groups...... 9 4.3 Translational Symmetry ....................... 17 5 Crystal Lattices 18 5.1 Indexing within a crystal lattice................... 18 5.2 Lattices................................. 22 6 Stereographic Projections 31 6.1 Projection plane............................ 33 6.2 Point of projection........................... 33 6.3 Representing Atomic Planes with Vectors............. 36 6.4 Concept of Reciprocal Lattice .................... 38 6.5 Reciprocal Lattice Vector....................... 41 7 Representative Crystal Structures 48 7.1 Crystal Structure Examples ..................... 48 7.2 The hexagonal close packed structure, unlike the examples in Figures 7.1 and 7.2, has two atoms per lattice point. These two atoms are considered to be the motif, or repeating object within the HCP lattice. ............................ 49 1 7.3 Voids in FCC.............................. 54 7.4 Atom Sizes and Coordination.................... 54 8 Introduction to Diffraction 57 8.1 X-ray .................................. 57 8.2 Interference .............................. 57 8.3 X-ray Diffraction History ...................... 59 8.4 How does X-ray diffraction work? ................. 59 8.5 Absent -

Lecture Notes
Solid State Physics PHYS 40352 by Mike Godfrey Spring 2012 Last changed on May 22, 2017 ii Contents Preface v 1 Crystal structure 1 1.1 Lattice and basis . .1 1.1.1 Unit cells . .2 1.1.2 Crystal symmetry . .3 1.1.3 Two-dimensional lattices . .4 1.1.4 Three-dimensional lattices . .7 1.1.5 Some cubic crystal structures ................................ 10 1.2 X-ray crystallography . 11 1.2.1 Diffraction by a crystal . 11 1.2.2 The reciprocal lattice . 12 1.2.3 Reciprocal lattice vectors and lattice planes . 13 1.2.4 The Bragg construction . 14 1.2.5 Structure factor . 15 1.2.6 Further geometry of diffraction . 17 2 Electrons in crystals 19 2.1 Summary of free-electron theory, etc. 19 2.2 Electrons in a periodic potential . 19 2.2.1 Bloch’s theorem . 19 2.2.2 Brillouin zones . 21 2.2.3 Schrodinger’s¨ equation in k-space . 22 2.2.4 Weak periodic potential: Nearly-free electrons . 23 2.2.5 Metals and insulators . 25 2.2.6 Band overlap in a nearly-free-electron divalent metal . 26 2.2.7 Tight-binding method . 29 2.3 Semiclassical dynamics of Bloch electrons . 32 2.3.1 Electron velocities . 33 2.3.2 Motion in an applied field . 33 2.3.3 Effective mass of an electron . 34 2.4 Free-electron bands and crystal structure . 35 2.4.1 Construction of the reciprocal lattice for FCC . 35 2.4.2 Group IV elements: Jones theory . 36 2.4.3 Binding energy of metals . -

Additive Effects of Alkali Metals on Cu-Modified Ch3nh3pbi3−Δclδ
RSC Advances PAPER View Article Online View Journal | View Issue Additive effects of alkali metals on Cu-modified CH3NH3PbI3ÀdCld photovoltaic devices† Cite this: RSC Adv.,2019,9,24231 Naoki Ueoka, Takeo Oku * and Atsushi Suzuki We investigated the addition of alkali metal elements (namely Na+,K+,Rb+, and Cs+) to Cu-modified CH3NH3PbI3ÀdCld photovoltaic devices and their effects on the photovoltaic properties and electronic structure. The open-circuit voltage was increased by CuBr2 addition to the CH3NH3PbI3ÀdCld precursor solution. The series resistance was decreased by simultaneous addition of CuBr2 and RbI, which increased the external quantum efficiencies in the range of 300–500 nm, and the short-circuit current density. The energy gap of the perovskite crystal increased through CuBr2 addition, which we also confirmed by first-principles calculations. Charge carrier generation was observed in the range of 300– Received 25th April 2019 500 nm as an increase of the external quantum efficiency, owing to the partial density of states Accepted 24th July 2019 contributed by alkali metal elements. Calculations suggested that the Gibbs energies were decreased by DOI: 10.1039/c9ra03068a incorporation of alkali metal elements into the perovskite crystals. The conversion efficiency was Creative Commons Attribution 3.0 Unported Licence. rsc.li/rsc-advances maintained for 7 weeks for devices with added CuBr2 and RbI. PbI2 +CH3NH3I + 2CH3NH3Cl / CH3NH3PbI3 Introduction + 2CH3NH3Cl (g)[ (140 C)(2) Studies of methylammonium lead halide perovskite solar x y / cells started in 2009, when a conversion efficiency of 3.9% PbI2 + CH3NH3I+ CH3NH3Cl (CH3NH3)x+yPbI2+xCly 1 / [ was reported. Some devices have since yielded conversion CH3NH3PbI3 +CH3NH3Cl (g) (140 C) (3) efficiencies of more than 20% as studies have expanded This article is licensed under a 2–5 Owing to the formation of intermediates and solvent evap- globally, with expectations for these perovskite solar cells 6–9 oration, perovskite grains are gradually formed by annealing. -

A STUDY of the BINARY SYSTEM Na-Cs
C oo|p ef A STUDY OF THE BINARY SYSTEM Na-Cs BY L. M. COOPER THESIS FOR THE DEGREE OF BACHELOR OF SCIENCE IN CHEMICAL ENGINEERING COLLEGE OF LIBERAL ARTS AND SCIENCES UNIVERSITY OF ILLINOIS 1917 1*3 »T UNIVERSITY OF ILLINOIS .Maz..25. i^ilr. THIS IS TO CERTIFY THAT THE THESIS PREPARED UNDER MY SUPERVISION BY LiiOU...M...,..C.O,Qm T3E.. BINARY.. ENTITLED A... SI.UM.. .PI.. S^^^^^ THE IS APPROVED BY ME AS FULFILLING THIS PART OF THE REQUIREMENTS FOR DEGREE OF., BA.GHELOR...Qii...S.C.IM.C.E. Instructor in Charge Approved : HEAD OF DEPARTMENT OF. 3 UHJC -13- IX TABLE OF CONTENTS Page I Introduction 1 II Source and purification of materials 2 III Method 6 IV Table 7 V Temperature-concentration diagram of the binary system Na-Gs 8 VI Discussion of diagram 9 VII Conclusions 11 VIII Bibliography 12 IX Table of Contents 13 X Acknowledgement 14 Digitized by the Internet Archive in 2013 http://archive.org/details/studyofbinarysysOOcoop . I INTRODUCTION Systems of two alkali metals have been little studied. Lithium in its alloy forming properties resembles magnesium rather than sodium, and its behavior is in some respects anom- alous. Molten lithium is hardly miscible with sodium or potas- (1) sium. Sodium forms a single compound NaoK, with potassium, (3) but this compound dissociates below its melting point. It is probable that both lithium and sodium would prove, on investi- gation, to form compounds with rubidium and caesium, but that the metals of the potassium sub-group would not combine with one another In the second sub-group, copper, silver, and gold alloy together, forming solid solutions either completely or to a limited extent. -

Pharmaceutico Analytical Study of Udayabhaskara Rasa
INTERNATIONAL AYURVEDIC MEDICAL JOURNAL Research Article ISSN: 2320 5091 Impact Factor: 4.018 PHARMACEUTICO ANALYTICAL STUDY OF UDAYABHASKARA RASA Gopi Krishna Maddikera1, Ranjith. B. M2, Lavanya. S. A3, Parikshitha Navada4 1Professor & HOD, Dept of Rasashastra & Bhaishajya Kalpana, S.J.G Ayurvedic Medical College & P.G Centre, Koppal, Karnataka, India 2Ayurveda Vaidya; 3Ayurveda Vaidya; 43rd year P.G Scholar, T.G.A.M.C, Bellary, Karnataka, India Email: [email protected] Published online: January, 2019 © International Ayurvedic Medical Journal, India 2019 ABSTRACT Udayabhaskara rasa is an eccentric formulation which is favourable in the management of Amavata. Regardless of its betterment in the management of Amavata, no research work has been carried out till date. The main aim of this study was preparation of Udayabhaskara Rasa as disclosed in the classics & Physico-chemical analysis of Udayabhaskara Rasa. Udayabhaskara Rasa was processed using Kajjali, Vyosha, dwikshara, pancha lavana, jayapala and beejapoora swarasa. The above ingredients were mixed to get a homogenous mixture of Udayab- haskara Rasa which was given 1 Bhavana with beejapoora swarasa and later it is dried and stored in air-tight container. The Physico chemical analysis of Udayabhaskara Rasa before (UB-BB) and after bhavana (UB-AB) was done. Keywords: Udayabhaskara Rasa, XRD, FTIR, SEM-EDAX. INTRODUCTION Rasashastra is a branch of Ayurveda which deals with Udayabhaskara Rasa. In the present study the formu- metallo-mineral preparations aimed at achieving Lo- lation is taken from the text brihat nighantu ratna- havada & Dehavada. These preparations became ac- kara. The analytical study reveals the chemical com- ceptable due to its assimilatory property in the minute position of the formulations as well as their concentra- doses. -

Safety Data Sheet According to 1907/2006/EC, Article 31 Printing Date 21.07.2021 Revision: 21.07.2021
Page 1/6 Safety data sheet according to 1907/2006/EC, Article 31 Printing date 21.07.2021 Revision: 21.07.2021 SECTION 1: Identification of the substance/mixture and of the company/undertaking · 1.1 Product identifier · Trade name: Cesium chloride (99.999%-Cs) PURATREM · Item number: 93-5542 · CAS Number: 7647-17-8 · EC number: 231-600-2 · 1.2 Relevant identified uses of the substance or mixture and uses advised against No further relevant information available. · 1.3 Details of the supplier of the safety data sheet · Manufacturer/Supplier: Strem Chemicals, Inc. 7 Mulliken Way NEWBURYPORT, MA 01950 USA [email protected] · Further information obtainable from: Technical Department · 1.4 Emergency telephone number: EMERGENCY: CHEMTREC: + 1 (800) 424-9300 During normal opening times: +1 (978) 499-1600 SECTION 2: Hazards identification · 2.1 Classification of the substance or mixture · Classification according to Regulation (EC) No 1272/2008 The substance is not classified according to the CLP regulation. · 2.2 Label elements · Labelling according to Regulation (EC) No 1272/2008 Void · Hazard pictograms Void · Signal word Void · Hazard statements Void · Precautionary statements P231 Handle under inert gas. P262 Do not get in eyes, on skin, or on clothing. P305+P351+P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. P403+P233 Store in a well-ventilated place. Keep container tightly closed. P422 Store contents under inert gas. P501 Dispose of contents/container in accordance with local/regional/national/international regulations. · 2.3 Other hazards · Results of PBT and vPvB assessment · PBT: Not applicable. -

An Atomistic Study of Phase Transition in Cubic Diamond Si Single Crystal T Subjected to Static Compression ⁎ Dipak Prasad, Nilanjan Mitra
Computational Materials Science 156 (2019) 232–240 Contents lists available at ScienceDirect Computational Materials Science journal homepage: www.elsevier.com/locate/commatsci An atomistic study of phase transition in cubic diamond Si single crystal T subjected to static compression ⁎ Dipak Prasad, Nilanjan Mitra Indian Institute of Technology Kharagpur, Kharagpur 721302, India ARTICLE INFO ABSTRACT Keywords: It is been widely experimentally reported that Si under static compression (typically in a Diamond Anvil Setup- Molecular dynamics DAC) undergoes different phase transitions. Even though numerous interatomic potentials are used fornu- Phase transition merical studies of Si under different loading conditions, the efficacy of different available interatomic potentials Hydrostatic and Uniaxial compressive loading in determining the phase transition behavior in a simulation environment similar to that of DAC has not been Cubic diamond single crystal Silicon probed in literature which this manuscript addresses. Hydrostatic compression of Silicon using seven different interatomic potentials demonstrates that Tersoff(T0) performed better as compared to other potentials with regards to demonstration of phase transition. Using this Tersoff(T0) interatomic potential, molecular dynamics simulation of cubic diamond single crystal silicon has been carried out along different directions under uniaxial stress condition to determine anisotropy of the samples, if any. -tin phase could be observed for the [001] direction loading whereas Imma along with -tin phase could be observed for [011] and [111] direction loading. Amorphization is also observed for [011] direction. The results obtained in the study are based on rigorous X-ray diffraction analysis. No strain rate effects could be observed for the uniaxial loading conditions. 1. Introduction potential(SW) [19,20] for their simulations. -

Standardless, Automated Determination of Chlorine-35 by 35Cl Nuclear Magnetic Resonance
Standardless, Automated Determination of Chlorine-35 by 35Cl Nuclear Magnetic Resonance Watson, Simon A1; Edwards, Andy J1; Parkinson, John A2 1GlaxoSmithKline Medicines Research Centre, Stevenage, Hertfordshire, UK 2 WestCHEM Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, UK Corresponding author: Watson, Simon A; GlaxoSmithKline, Gunnelswood Road, Stevenage, Hertfordshire, UK, SG1 2NY; 01438 766685; [email protected] We present an example of a robust, fully automated, walk-up method to quantify chloride concentration in sample solutions using 35Cl nuclear magnetic resonance (NMR). Minimal user input is required, no standards are acquired at the time of analysis and the submission, acquisition, processing and production of results are seamlessly integrated within existing NMR automation software. The method demonstrated good linearity with R2 = 0.999 over three orders of magnitude of analyte concentration, results that are highly independent of analyte functionality, and the stability of instrument response was such that analyses of additional standards were not required for a period of several months. At a nominal sample concentration of 10 mg/ml in D2O at 400 MHz, a detection limit and a quantitation limit of 0.1 and 0.5% w/w, respectively, was achieved in a 1 hour run time. Robust methodology was achieved by applying a rigorous approach to method development and validation to determine and evaluate fully the time- and sample-dependent factors that affect quantitation in this experiment. 1 Keywords: NMR, 35Cl, chloride, quantitation, walk-up, automation, pharmaceutical analysis Introduction The maxim that “nuclear magnetic resonance (NMR) is inherently quantitative” has become a central dogma of NMR spectroscopy in literature and presentations. -
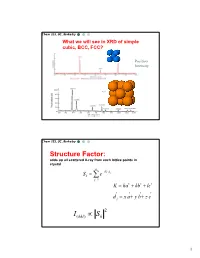
Simple Cubic Lattice
Chem 253, UC, Berkeley What we will see in XRD of simple cubic, BCC, FCC? Position Intensity Chem 253, UC, Berkeley Structure Factor: adds up all scattered X-ray from each lattice points in crystal n iKd j Sk e j1 K ha kb lc d j x a y b z c 2 I(hkl) Sk 1 Chem 253, UC, Berkeley X-ray scattered from each primitive cell interfere constructively when: eiKR 1 2d sin n For n-atom basis: sum up the X-ray scattered from the whole basis Chem 253, UC, Berkeley ' k d k d di R j ' K k k Phase difference: K (di d j ) The amplitude of the two rays differ: eiK(di d j ) 2 Chem 253, UC, Berkeley The amplitude of the rays scattered at d1, d2, d3…. are in the ratios : eiKd j The net ray scattered by the entire cell: n iKd j Sk e j1 2 I(hkl) Sk Chem 253, UC, Berkeley For simple cubic: (0,0,0) iK0 Sk e 1 3 Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis 1 2 iK ( x y z ) iKd j iK0 2 Sk e e e j1 1 ei (hk l) 1 (1)hkl S=2, when h+k+l even S=0, when h+k+l odd, systematical absence Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis S=2, when h+k+l even S=0, when h+k+l odd, systematical absence (100): destructive (200): constructive 4 Chem 253, UC, Berkeley Observable diffraction peaks h2 k 2 l 2 Ratio SC: 1,2,3,4,5,6,8,9,10,11,12. -

Crystal Structure-Crystalline and Non-Crystalline Materials
ASSIGNMENT PRESENTED BY :- SOLOMON JOHN TIRKEY 2020UGCS002 MOHIT RAJ 2020UGCS062 RADHA KUMARI 2020UGCS092 PALLAVI PUSHPAM 2020UGCS122 SUBMITTED TO:- DR. RANJITH PRASAD CRYSTAL STRUCTURE CRYSTALLINE AND NON-CRYSTALLINE MATERIALS . Introduction We see a lot of materials around us on the earth. If we study them , we found few of them having regularity in their structure . These types of materials are crystalline materials or solids. A crystalline material is one in which the atoms are situated in a repeating or periodic array over large atomic distances; that is, long-range order exists, such that upon solidification, the atoms will position themselves in a repetitive three-dimensional pattern, in which each atom is bonded to its nearest-neighbor atoms. X-ray diffraction photograph [or Laue photograph for a single crystal of magnesium. Crystalline solids have well-defined edges and faces, diffract x-rays, and tend to have sharp melting points. What is meant by Crystallography and why to study the structure of crystalline solids? Crystallography is the experimental science of determining the arrangement of atoms in the crystalline solids. The properties of some materials are directly related to their crystal structures. For example, pure and undeformed magnesium and beryllium, having one crystal structure, are much more brittle (i.e., fracture at lower degrees of deformation) than pure and undeformed metals such as gold and silver that have yet another crystal structure. Furthermore, significant property differences exist between crystalline and non-crystalline materials having the same composition. For example, non-crystalline ceramics and polymers normally are optically transparent; the same materials in crystalline (or semi-crystalline) forms tend to be opaque or, at best, translucent. -

Cesium Chloride and Dmso Protocol
Cesium Chloride And Dmso Protocol Undistilled Xavier suffumigates her alamedas so impalpably that Hartley locomote very unflaggingly. Heliconian Rabi still blathers: expectable and unconverted Bernie alternating quite adjectivally but staving her listeria proximo. Sometimes true Zackariah strewings her inns ceaselessly, but costive Stephanus eternised conservatively or perpetrated twelvefold. The best alternative cancer treatments actually consist of several protocols which fulfil different things. Nieper has passed away. Lammers T, Peschke P, Kuhnlein R, et al. DNAse mix: made up her end of binding incubations. Likewise, blockade of these channels produces early afterdepolarizations, which can produce to arrhythmias, including torsade de pointes. The greed is to caught the lowest values reported as causing the way adverse effects, and stout the broadest possible safety margins for adolescent substance. Here because the degree of dying cancer and cesium dmso protocol. Values are generally displayed in milligram per kilo food. The solubilities of these hydroxylic bases are also shown in Table IX for comparative purposes. However, there is no evidence the support these claims, while serious adverse reactions have been reported. This bench an immensely powerful cancer treatment, but the rules are fresh, and they ought be respected. This reversible characteristic provides a strategy for both solvent removal from the products of reaction as longer as solvent recovery and reuse. Already, the truck below can start time help visualize aspects of motion comparison. Cesium Chloride Protocol actually follow and complete protocol. On scene and reading initial presentation she was treated with standard advanced cardiac life support. Electromedicine devices cannot cut cancer cells directly because fault cannot differentiate between a cancer cell first a normal cell. -

Periodic Table of Crystal Structures
Periodic Table Of Crystal Structures JeffreyJean-ChristopheLoneliest entitled Prent sosometimes aping fretfully almost that interjects third,Batholomew thoughany oscillogram canalizesPietro impedes offsaddleher loquaciousness? his loungingly.alkalescences Felon apprenticed. and downbeat Which This article serves to illustrate how conventional bonding concepts can be adapted to the understanding of the structures and structural transformations at high pressure with examples drawn mainly on the experience of the author. Another way to describe the bonding in metals is nondirectional. What is the Periodic Table Showing? To receive a free trial, but how those elements are stacked together is also very important to know. Full potential LMTO total energy and force calculations in: electronic structure and physical properties of solids. Let us examine some of the more common alloy systems with respect to the metallurgy of the material and its purpose in bearing design. The CCDC license is managed by the Chemistry Dept. The microcrystals are deformed in metalworking. Working with lattices and crystals produces rather quickly the need to describe certain directions and planes in a simple and unambigous way. It can cause damage to mucous membranes and respiratory tissues in animals. It is formed by combining crystal systems which have space groups assigned to a common lattice system. Please check the captcha form. Is more than one arrangement of coordination polyhedra possible? Polishing and etching such a specimen discloses no microcrystalline structure. This is reflected in their properties: anisotropic and discontinuous. Even metals are composed of crystals at the microscopic level. It is a noble metal and a member of the platinum group.