Integrative Annotation and Knowledge Discovery of Kinase Post
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

The Drug Sensitivity and Resistance Testing (DSRT) Approach
A phenotypic screening and machine learning platform eciently identifies triple negative breast cancer-selective and readily druggable targets Prson Gautam 1 Alok Jaiswal 1 Tero Aittokallio 1, 2 Hassan Al Ali 3 Krister Wennerberg 1,4 Identifying eective oncogenic targets is challenged by the complexity of genetic alterations in 1Institute for Molecular Medicine Finland (FIMM), HiLIFE, University of Helsinki, Finland cancer and their poorly understood relation to cell function and survival. There is a need for meth- Current kinome coverage of kinase inhibitors in TNBC exhibit diverse kinase dependencies MFM-223 is selectively addicted to FGFR2 2Department of Mathematics and Statistics, University of Turku, Finland 3The Miami Project to Cure Paralysis, Peggy and Harold Katz Family Drug Discovery Center, A A Sylvester Comprehensive Cancer Center, and Department of Neurological Surgery and Medicine ods that rapidly and accurately identify “pharmacologically eective” targets without the require- clinical evaluation TN Kinases MFM-223 CAL-120 MDA-MB-231 TNBC TNBC TNBC TNBC TNBC TNBC HER2+ 100 University of Miami Miller School of Medicine, Miami, FL 33136, USA. non- HER2+ FGFR1 0.97 0.00 0.00 MFM-223 BL1 BL2 M MSL IM LAR ER+, PR+ 50 ment for priori knowledge of complex signaling networks. We developed an approach that uses ma- cancerous FGFR2 56.46 0.00 0.00 CAL-120 25 4 MDA-MB-231 Biotech Research & Innovation Centre (BRIC) and Novo Nordisk Foundation Center HCC1937 CAL-85-1 CAL-120 MDA-MB-231 DU4475 CAL-148 MCF-10A SK-BR-3 BT-474 FGFR3 25.10 0.00 0.00 0 chine learning to relate results from unbiased phenotypic screening of kinase inhibitors to their bio- for Stem Cell Biology (DanStem), University of Copenhagen, Denmark HCC1599 HDQ-P1 BT-549 MDA-MB-436 MFM-223 FGFR4 0.00 0.00 0.00 MAXIS*Bk Clinical status MDA-MB-468 CAL-51 Hs578T MDA-MB-453 score chemical activity data. -

The Prevalence of MADH4 and BMPR1A Mutations in Juvenile Polyposis and Absence of BMPR2, BMPR1B, and ACVR1 Mutations
484 ORIGINAL ARTICLE J Med Genet: first published as 10.1136/jmg.2004.018598 on 2 July 2004. Downloaded from The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations J R Howe, M G Sayed, A F Ahmed, J Ringold, J Larsen-Haidle, A Merg, F A Mitros, C A Vaccaro, G M Petersen, F M Giardiello, S T Tinley, L A Aaltonen, H T Lynch ............................................................................................................................... J Med Genet 2004;41:484–491. doi: 10.1136/jmg.2004.018598 Background: Juvenile polyposis (JP) is an autosomal dominant syndrome predisposing to colorectal and gastric cancer. We have identified mutations in two genes causing JP, MADH4 and bone morphogenetic protein receptor 1A (BMPR1A): both are involved in bone morphogenetic protein (BMP) mediated signalling and are members of the TGF-b superfamily. This study determined the prevalence of mutations See end of article for in MADH4 and BMPR1A, as well as three other BMP/activin pathway candidate genes in a large number authors’ affiliations ....................... of JP patients. Methods: DNA was extracted from the blood of JP patients and used for PCR amplification of each exon of Correspondence to: these five genes, using primers flanking each intron–exon boundary. Mutations were determined by Dr J R Howe, Department of Surgery, 4644 JCP, comparison to wild type sequences using sequence analysis software. A total of 77 JP cases were University of Iowa College sequenced for mutations in the MADH4, BMPR1A, BMPR1B, BMPR2, and/or ACVR1 (activin A receptor) of Medicine, 200 Hawkins genes. The latter three genes were analysed when MADH4 and BMPR1A sequencing found no mutations. -

Casein Kinase 1 Isoforms in Degenerative Disorders
CASEIN KINASE 1 ISOFORMS IN DEGENERATIVE DISORDERS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Theresa Joseph Kannanayakal, M.Sc., M.S. * * * * * The Ohio State University 2004 Dissertation Committee: Approved by Professor Jeff A. Kuret, Adviser Professor John D. Oberdick Professor Dale D. Vandre Adviser Professor Mike X. Zhu Biophysics Graduate Program ABSTRACT Casein Kinase 1 (CK1) enzyme is one of the largest family of Serine/Threonine protein kinases. CK1 has a wide distribution spanning many eukaryotic families. In cells, its kinase activity has been found in various sub-cellular compartments enabling it to phosphorylate many proteins involved in cellular maintenance and disease pathogenesis. Tau is one such substrate whose hyperphosphorylation results in degeneration of neurons in Alzheimer’s disease (AD). AD is a slow neuroprogessive disorder histopathologically characterized by Granulovacuolar degeneration bodies (GVBs) and intraneuronal accumulation of tau in Neurofibrillary Tangles (NFTs). The level of CK1 isoforms, CK1α, CK1δ and CK1ε has been shown to be elevated in AD. Previous studies of the correlation of CK1δ with lesions had demonstrated its importance in tau hyperphosphorylation. Hence we investigated distribution of CK1α and CK1ε with the lesions to understand if they would play role in tau hyperphosphorylation similar to CK1δ. The kinase results were also compared with lesion correlation studies of peptidyl cis/trans prolyl isomerase (Pin1) and caspase-3. Our results showed that among the enzymes investigated, CK1 isoforms have the greatest extent of colocalization with the lesions. We have also investigated the distribution of CK1α with different stages of NFTs that follow AD progression. -

3De8d416-B844-4E9a-B3fb-A2e883a4083f 11119
Edinburgh Research Explorer Last rolls of the yoyo: Assessing the human canonical protein count Citation for published version: Southan, C 2017, 'Last rolls of the yoyo: Assessing the human canonical protein count', F1000Research, vol. 6, pp. 448+. https://doi.org/10.12688/f1000research.11119.1 Digital Object Identifier (DOI): 10.12688/f1000research.11119.1 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: F1000Research Publisher Rights Statement: This is an open access article distributed under the terms of the Creative Commons Attribution Licence, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Data associated with the article are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication). General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately -

Study of Conditions for the Emergence of Cellular Communication Using Self-Adaptive Multi-Agent Systems Sebastien Maignan
Study of conditions for the emergence of cellular communication using self-adaptive multi-agent systems Sebastien Maignan To cite this version: Sebastien Maignan. Study of conditions for the emergence of cellular communication using self- adaptive multi-agent systems. Artificial Intelligence [cs.AI]. Université Paul Sabatier - Toulouse III, 2018. English. NNT : 2018TOU30094. tel-02094368 HAL Id: tel-02094368 https://tel.archives-ouvertes.fr/tel-02094368 Submitted on 9 Apr 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Délivré par l'Université Toulouse 3 Paul Sabatier (UT3 Paul Sabatier) Sébastien Maignan Le 30 août 2018 Study of conditions for the emergence of cellular communication using self-adaptive multi-agent systems École doctorale et discipline ou spécialité ED MITT : Domaine STIC : Intelligence Artificielle Unité de recherche Institut de Recherche en Informatique de Toulouse Directrice(s) ou Directeur(s) de Thèse Pierre Glize, Carole Bernon Jury Marie BEURTON-AIMAR Maître de Conférences, HdR, Université de Bordeaux Rapporteur Vincent -

Bromodomain Protein BRDT Directs ΔNp63 Function and Super
Cell Death & Differentiation (2021) 28:2207–2220 https://doi.org/10.1038/s41418-021-00751-w ARTICLE Bromodomain protein BRDT directs ΔNp63 function and super-enhancer activity in a subset of esophageal squamous cell carcinomas 1 1 2 1 1 2 Xin Wang ● Ana P. Kutschat ● Moyuru Yamada ● Evangelos Prokakis ● Patricia Böttcher ● Koji Tanaka ● 2 3 1,3 Yuichiro Doki ● Feda H. Hamdan ● Steven A. Johnsen Received: 25 August 2020 / Revised: 3 February 2021 / Accepted: 4 February 2021 / Published online: 3 March 2021 © The Author(s) 2021. This article is published with open access Abstract Esophageal squamous cell carcinoma (ESCC) is the predominant subtype of esophageal cancer with a particularly high prevalence in certain geographical regions and a poor prognosis with a 5-year survival rate of 15–25%. Despite numerous studies characterizing the genetic and transcriptomic landscape of ESCC, there are currently no effective targeted therapies. In this study, we used an unbiased screening approach to uncover novel molecular precision oncology targets for ESCC and identified the bromodomain and extraterminal (BET) family member bromodomain testis-specific protein (BRDT) to be 1234567890();,: 1234567890();,: uniquely expressed in a subgroup of ESCC. Experimental studies revealed that BRDT expression promotes migration but is dispensable for cell proliferation. Further mechanistic insight was gained through transcriptome analyses, which revealed that BRDT controls the expression of a subset of ΔNp63 target genes. Epigenome and genome-wide occupancy studies, combined with genome-wide chromatin interaction studies, revealed that BRDT colocalizes and interacts with ΔNp63 to drive a unique transcriptional program and modulate cell phenotype. Our data demonstrate that these genomic regions are enriched for super-enhancers that loop to critical ΔNp63 target genes related to the squamous phenotype such as KRT14, FAT2, and PTHLH. -

PIR Brochure
Protein Information Resource Integrated Protein Informatics Resource for Genomic & Proteomic Research For four decades the Protein Information Resource (PIR) has provided databases and protein sequence analysis tools to the scientific community, including the Protein Sequence Database, which grew out from the Atlas of Protein Sequence and Structure, edited by Margaret Dayhoff [1965-1978]. Currently, PIR major activities include: i) UniProt (Universal Protein Resource) development, ii) iProClass protein data integration and ID mapping, iii) PIRSF protein pir.georgetown.edu classification, and iv) iProLINK protein literature mining and ontology development. UniProt – Universal Protein Resource What is UniProt? UniProt is the central resource for storing and UniProt (Universal Protein Resource) http://www.uniprot.org interconnecting information from large and = + + disparate sources and the most UniProt: the world's most comprehensive catalog of information on proteins comprehensive catalog of protein sequence and functional annotation. UniProt Knowledgebase UniProt Reference UniProt Archive (UniProtKB) Clusters (UniRef) (UniParc) When to use UniProt databases Integration of Swiss-Prot, TrEMBL Non-redundant reference A stable, and PIR-PSD sequences clustered from comprehensive Use UniProtKB to retrieve curated, reliable, Fully classified, richly and accurately UniProtKB and UniParc for archive of all publicly annotated protein sequences with comprehensive or fast available protein comprehensive information on proteins. minimal redundancy and extensive sequence searches at 100%, sequences for Use UniRef to decrease redundancy and cross-references 90%, or 50% identity sequence tracking from: speed up sequence similarity searches. TrEMBL section UniRef100 Swiss-Prot, Computer-annotated protein sequences TrEMBL, PIR-PSD, Use UniParc to access to archived sequences EMBL, Ensembl, IPI, and their source databases. -
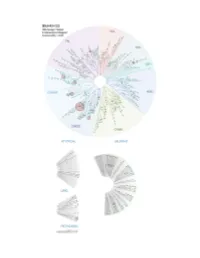
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -

Loss of BCL-3 Sensitises Colorectal Cancer Cells to DNA Damage, Revealing A
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.03.454995; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Loss of BCL-3 sensitises colorectal cancer cells to DNA damage, revealing a role for BCL-3 in double strand break repair by homologous recombination Authors: Christopher Parker*1, Adam C Chambers*1, Dustin Flanagan2, Tracey J Collard1, Greg Ngo3, Duncan M Baird3, Penny Timms1, Rhys G Morgan4, Owen Sansom2 and Ann C Williams1. *Joint first authors. Author affiliations: 1. Colorectal Tumour Biology Group, School of Cellular and Molecular Medicine, Faculty of Life Sciences, Biomedical Sciences Building, University Walk, University of Bristol, Bristol, BS8 1TD, UK 2. Cancer Research UK Beatson Institute, Garscube Estate, Switchback Road, Bearsden Glasgow, G61 1BD UK 3. Division of Cancer and Genetics, School of Medicine, Cardiff University, Cardiff, CF14 4XN UK 4. School of Life Sciences, University of Sussex, Sussex House, Falmer, Brighton, BN1 9RH UK 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.08.03.454995; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract (250 words) Objective: The proto-oncogene BCL-3 is upregulated in a subset of colorectal cancers (CRC) and increased expression of the gene correlates with poor patient prognosis. The aim is to investigate whether inhibiting BCL-3 can increase the response to DNA damage in CRC. -

BRDT Is an Essential Epigenetic Regulator for Proper Chromatin Organization, Silencing of Sex Chromosomes and Crossover Formation in Male Meiosis
RESEARCH ARTICLE BRDT is an essential epigenetic regulator for proper chromatin organization, silencing of sex chromosomes and crossover formation in male meiosis Marcia Manterola1,2, Taylor M. Brown1, Min Young Oh1, Corey Garyn1, Bryan J. Gonzalez3, Debra J. Wolgemuth1,3,4* a1111111111 1 Department of Genetics & Development, Columbia University Medical Center, New York, NY, United States of America, 2 Human Genetics Program, Institute of Biomedical Sciences, Faculty of Medicine, a1111111111 University of Chile, Santiago, Chile, 3 Institute of Human Nutrition, Columbia University Medical Center, New a1111111111 York, NY,United States of America, 4 Department of Obstetrics & Gynecology, Columbia University Medical a1111111111 Center, New York, NY,United States of America a1111111111 * [email protected] Abstract OPEN ACCESS Citation: Manterola M, Brown TM, Oh MY, Garyn The double bromodomain and extra-terminal domain (BET) proteins are critical epigenetic C, Gonzalez BJ, Wolgemuth DJ (2018) BRDT is an readers that bind to acetylated histones in chromatin and regulate transcriptional activity essential epigenetic regulator for proper chromatin and modulate changes in chromatin structure and organization. The testis-specific BET organization, silencing of sex chromosomes and member, BRDT, is essential for the normal progression of spermatogenesis as mutations in crossover formation in male meiosis. PLoS Genet 14(3): e1007209. https://doi.org/10.1371/journal. the Brdt gene result in complete male sterility. Although BRDT is expressed in both sper- pgen.1007209 matocytes and spermatids, loss of the first bromodomain of BRDT leads to severe defects Editor: P. Jeremy Wang, University of in spermiogenesis without overtly compromising meiosis. In contrast, complete loss of Pennsylvania, UNITED STATES BRDT blocks the progression of spermatocytes into the first meiotic division, resulting in a Received: April 25, 2017 complete absence of post-meiotic cells.