Heritability Estimates of Complex Intelligence and Associated Genetics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lineage-Specific Mapping of Quantitative Trait Loci
Heredity (2013) 111, 106–113 & 2013 Macmillan Publishers Limited All rights reserved 0018-067X/13 www.nature.com/hdy ORIGINAL ARTICLE Lineage-specific mapping of quantitative trait loci C Chen1 and K Ritland We present an approach for quantitative trait locus (QTL) mapping, termed as ‘lineage-specific QTL mapping’, for inferring allelic changes of QTL evolution along with branches in a phylogeny. We describe and analyze the simplest case: by adding a third taxon into the normal procedure of QTL mapping between pairs of taxa, such inferences can be made along lineages to a presumed common ancestor. Although comparisons of QTL maps among species can identify homology of QTLs by apparent co- location, lineage-specific mapping of QTL can classify homology into (1) orthology (shared origin of QTL) versus (2) paralogy (independent origin of QTL within resolution of map distance). In this light, we present a graphical method that identifies six modes of QTL evolution in a three taxon comparison. We then apply our model to map lineage-specific QTLs for inbreeding among three taxa of yellow monkey-flower: Mimulus guttatus and two inbreeders M. platycalyx and M. micranthus, but critically assuming outcrossing was the ancestral state. The two most common modes of homology across traits were orthologous (shared ancestry of mutation for QTL alleles). The outbreeder M. guttatus had the fewest lineage-specific QTL, in accordance with the presumed ancestry of outbreeding. Extensions of lineage-specific QTL mapping to other types of data and crosses, and to inference of ancestral QTL state, are discussed. Heredity (2013) 111, 106–113; doi:10.1038/hdy.2013.24; published online 24 April 2013 Keywords: Quantitative trait locus mapping; evolution of mating systems; Mimulus INTRODUCTION common ancestor of these two taxa, back to the common ancestor of Knowledge of the genetic architecture of complex traits can offer all three taxa and forwards again to the third taxon. -

Heritability in the Era of Molecular Genetics: Some Thoughts for Understanding Genetic Influences on Behavioural Traits
European Journal of Personality, Eur. J. Pers. 25: 254–266 (2011) Published online (wileyonlinelibrary.com) DOI: 10.1002/per.836 Heritability in the Era of Molecular Genetics: Some Thoughts for Understanding Genetic Influences on Behavioural Traits WENDY JOHNSON1,2*, LARS PENKE1 and FRANK M. SPINATH3 1Centre for Cognitive Ageing and Cognitive Epidemiology and Department of Psychology, University of Edinburgh, Edinburgh, UK 2Department of Psychology, University of Minnesota, Minneapolis, Minnesota, USA 3Department of Psychology, Saarland University, Saarbruecken, Germany Abstract: Genetic influences on behavioural traits are ubiquitous. When behaviourism was the dominant paradigm in psychology, demonstrations of heritability of behavioural and psychological constructs provided important evidence of its limitations. Now that genetic influences on behavioural traits are generally accepted, we need to recognise the limitations of heritability as an indicator of both the aetiology and likelihood of discovering molecular genetic associations with behavioural traits. We review those limitations and conclude that quantitative genetics and genetically informative research designs are still critical to understanding the roles of gene‐environment interplay in developmental processes, though not necessarily in the ways commonly discussed. Copyright © 2011 John Wiley & Sons, Ltd. Key words: genetic influences; twin study; heritability Much is often made of new findings of the presence of environmental influences but emphasised that these genetic influences -

Interpreting Noncoding Genetic Variation in Complex Traits and Human Disease
REVIEW Interpreting noncoding genetic variation in complex traits and human disease Lucas D Ward1,2 & Manolis Kellis1,2 Association studies provide genome-wide information about the genetic basis of complex disease, but medical research has focused primarily on protein-coding variants, owing to the difficulty of interpreting noncoding mutations. This picture has changed with advances in the systematic annotation of functional noncoding elements. Evolutionary conservation, functional genomics, chromatin state, sequence motifs and molecular quantitative trait loci all provide complementary information about the function of noncoding sequences. These functional maps can help with prioritizing variants on risk haplotypes, filtering mutations encountered in the clinic and performing systems-level analyses to reveal processes underlying disease associations. Advances in predictive modeling can enable data-set integration to reveal pathways shared across loci and alleles, and richer regulatory models can guide the search for epistatic interactions. Lastly, new massively parallel reporter experiments can systematically validate regulatory predictions. Ultimately, advances in regulatory and systems genomics can help unleash the value of whole-genome sequencing for personalized genomic risk assessment, diagnosis and treatment. Understanding the genetic basis of disease can transform medicine ture of the human genome, and its systematic mapping by the HapMap by elucidating relevant biochemical pathways for drug targets and by Project9, allowed single-nucleotide polymorphisms (SNPs) to be used enabling personalized risk assessments1,2. With the evolution of technol- as markers for common haplotypes, which could be genotyped using ogies over the past century, geneticists are no longer limited to studying chip technology. The stage was set for a flood of unbiased, genome-wide Mendelian disorders and can tackle complex phenotypes. -
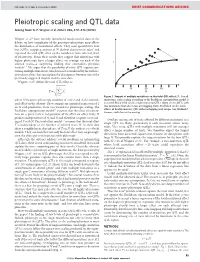
Pleiotropic Scaling and QTL Data Arising From: G
NATURE | Vol 456 | 4 December 2008 BRIEF COMMUNICATIONS ARISING Pleiotropic scaling and QTL data Arising from: G. P. Wagner et al. Nature 452, 470–473 (2008) Wagner et al.1 have recently introduced much-needed data to the debate on how complexity of the genotype–phenotype map affects the distribution of mutational effects. They used quantitative trait loci (QTLs) mapping analysis of 70 skeletal characters in mice2 and regressed the total QTL effect on the number of traits affected (level of pleiotropy). From their results they suggest that mutations with higher pleiotropy have a larger effect, on average, on each of the T affected traits—a surprising finding that contradicts previous models3–7. We argue that the possibility of some QTL regions con- taining multiple mutations, which was not considered by the authors, introduces a bias that can explain the discrepancy between one of the previously suggested models and the new data. Wagner et al.1 define the total QTL effect as vffiffiffiffiffiffiffiffiffiffiffiffiffi u uXN 10 20 30 ~t 2 T Ai N i~1 Figure 1 | Impact of multiple mutations on the total QTL effect, T. For all where N measures pleiotropy (number of traits) and Ai the normal- mutations, strict scaling according to the Euclidean superposition model is ized effect on the ith trait. They compare an empirical regression of T assumed. Black filled circles, single-mutation QTLs. Open circles, QTLs with on N with predictions from two models for pleiotropic scaling. The two mutations that affect non-overlapping traits. Red filled circles, total Euclidean superposition model3,4 assumes that the effect of a muta- effects of double-mutant QTL with overlapping trait ranges (see Methods). -

Transformations of Lamarckism Vienna Series in Theoretical Biology Gerd B
Transformations of Lamarckism Vienna Series in Theoretical Biology Gerd B. M ü ller, G ü nter P. Wagner, and Werner Callebaut, editors The Evolution of Cognition , edited by Cecilia Heyes and Ludwig Huber, 2000 Origination of Organismal Form: Beyond the Gene in Development and Evolutionary Biology , edited by Gerd B. M ü ller and Stuart A. Newman, 2003 Environment, Development, and Evolution: Toward a Synthesis , edited by Brian K. Hall, Roy D. Pearson, and Gerd B. M ü ller, 2004 Evolution of Communication Systems: A Comparative Approach , edited by D. Kimbrough Oller and Ulrike Griebel, 2004 Modularity: Understanding the Development and Evolution of Natural Complex Systems , edited by Werner Callebaut and Diego Rasskin-Gutman, 2005 Compositional Evolution: The Impact of Sex, Symbiosis, and Modularity on the Gradualist Framework of Evolution , by Richard A. Watson, 2006 Biological Emergences: Evolution by Natural Experiment , by Robert G. B. Reid, 2007 Modeling Biology: Structure, Behaviors, Evolution , edited by Manfred D. Laubichler and Gerd B. M ü ller, 2007 Evolution of Communicative Flexibility: Complexity, Creativity, and Adaptability in Human and Animal Communication , edited by Kimbrough D. Oller and Ulrike Griebel, 2008 Functions in Biological and Artifi cial Worlds: Comparative Philosophical Perspectives , edited by Ulrich Krohs and Peter Kroes, 2009 Cognitive Biology: Evolutionary and Developmental Perspectives on Mind, Brain, and Behavior , edited by Luca Tommasi, Mary A. Peterson, and Lynn Nadel, 2009 Innovation in Cultural Systems: Contributions from Evolutionary Anthropology , edited by Michael J. O ’ Brien and Stephen J. Shennan, 2010 The Major Transitions in Evolution Revisited , edited by Brett Calcott and Kim Sterelny, 2011 Transformations of Lamarckism: From Subtle Fluids to Molecular Biology , edited by Snait B. -

1 ROBERT PLOMIN on Behavioral Genetics Genetic Links Common
Genetic links • Background – Why has educational psychology ben so slow ROBERT PLOMIN to accept genetics? on Behavioral Genetics • Beyond nature vs. nurture – 3 findings from genetic research with far- reaching implications for education: October, 2004 The nature of nurture The abnormal is normal Generalist genes • Molecular Genetics Quantitative genetics: twin and adoption studies Common medical disorders Identical twins Fraternal twins (monozygotic, MZ) (dizygotic, DZ) 1 Twin concordances in TEDS at 7 years Twins Early Development Study (TEDS) for low reading and maths Perceptions of nature/nurture: % parents (N=1340) and teachers Beyond nature vs. nurture: (N=667) indicating that genes are at least as important as environment (Walker & Plomin, Ed. Psych., in press) The nature of nurture • Genetic influence on experience • Genotype-environment correlation – Children’s genetic propensities are correlated with their experiences passive evocative active 2 Beyond nature vs. nurture: Genotype-environment correlation The nature of nurture • Passive • Genotype-environment correlation – e.g., family background (SES) and school – Children’s genetic propensities are correlated with achievement their experiences passive • Evocative evocative – Children evoke experiences for genetic reasons Active • Active • Molecular genetics – Children actively create environments that foster – Find genes associated with learning experiences their genetic propensities Beyond nature vs. nurture: The one-gene one-disability (OGOD) model The abnormal is normal • Common learning disabilities are the quantitative extreme of the same genetic and environmental factors responsible for normal variation in learning abilities. • There are no common learning disabilities, just the extremes of normal variation. 3 OGOD (rare) and QTL (common) disorders The quantitative trait locus (QTL) model Beyond nature vs. -

High Heritability of Telomere Length, but Low Evolvability, and No Significant
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.16.423128; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 High heritability of telomere length, but low evolvability, and no significant 2 heritability of telomere shortening in wild jackdaws 3 4 Christina Bauch1*, Jelle J. Boonekamp1,2, Peter Korsten3, Ellis Mulder1, Simon Verhulst1 5 6 Affiliations: 7 1 Groningen Institute for Evolutionary Life Sciences, University of Groningen, Nijenborgh 7, 8 9747AG Groningen, The Netherlands 9 2 present address: Institute of Biodiversity Animal Health & Comparative Medicine, College 10 of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow G12 8QQ, United 11 Kingdom 12 3 Department of Animal Behaviour, Bielefeld University, Morgenbreede 45, 33615 Bielefeld, 13 Germany 14 15 16 * Correspondence to: 17 [email protected] 18 19 Running head: High heritability of telomere length bioRxiv preprint doi: https://doi.org/10.1101/2020.12.16.423128; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 20 Abstract 21 Telomere length (TL) and shortening rate predict survival in many organisms. Evolutionary 22 dynamics of TL in response to survival selection depend on the presence of genetic variation 23 that selection can act upon. However, the amount of standing genetic variation is poorly known 24 for both TL and TL shortening rate, and has not been studied for both traits in combination in 25 a wild vertebrate. -

Genetics in Harry Potter's World: Lesson 2
Genetics in Harry Potter’s World Lesson 2 • Beyond Mendelian Inheritance • Genetics of Magical Ability 1 Rules of Inheritance • Some traits follow the simple rules of Mendelian inheritance of dominant and recessive genes. • Complex traits follow different patterns of inheritance that may involve multiples genes and other factors. For example, – Incomplete or blended dominance – Codominance – Multiple alleles – Regulatory genes Any guesses on what these terms may mean? 2 Incomplete Dominance • Incomplete dominance results in a phenotype that is a blend of a heterozygous allele pair. Ex., Red flower + Blue flower => Purple flower • If the dragons in Harry Potter have fire-power alleles F (strong fire) and F’ (no fire) that follow incomplete dominance, what are the phenotypes for the following dragon-fire genotypes? – FF – FF’ – F’F’ 3 Incomplete Dominance • Incomplete dominance results in a phenotype that is a blend of the two traits in an allele pair. Ex., Red flower + Blue flower => Purple flower • If the Dragons in Harry Potter have fire-power alleles F (strong fire) and F’ (no fire) that follow incomplete dominance, what are the phenotypes for the following dragon-fire genotypes: Genotypes Phenotypes FF strong fire FF’ moderate fire (blended trait) F’F’ no fire 4 Codominance • Codominance results in a phenotype that shows both traits of an allele pair. Ex., Red flower + White flower => Red & White spotted flower • If merpeople have tail color alleles B (blue) and G (green) that follow the codominance inheritance rule, what are possible genotypes and phenotypes? Genotypes Phenotypes 5 Codominance • Codominance results in a phenotype that shows both traits of an allele pair. -

The Genetics of Complex Traits Programme Course 7.5 Credits Genetiska Mekanismer Bakom Komplexa Egenskaper 8MEA14 Valid From: 2019 Autumn Semester
DNR LIU-2019-00662 1(5) The genetics of complex traits Programme course 7.5 credits Genetiska mekanismer bakom komplexa egenskaper 8MEA14 Valid from: 2019 Autumn semester Determined by The Board for First and Second Cycle Programmes at the Faculty of Medicine and Health Sciences Date determined 2019-03-07 LINKÖPING UNIVERSITY FACULTY OF MEDICINE AND HEALTH SCIENCES LINKÖPING UNIVERSITY THE GENETICS OF COMPLEX TRAITS FACULTY OF MEDICINE AND HEALTH SCIENCES 2(5) Main field of study Medical Biology Course level Second cycle Advancement level A1X Course offered for Master's Programme in Experimental and Medical Biosciences Entry requirements Degree of Bachelor of Science, 180 ECTS in a major subject area such as medicine, biology, technology/natural sciences, odontology or veterinary medicine. 90 ECTS of courses included in the Bachelor degree should be in subjects such as biochemistry, cell biology, molecular biology, genetics, gene technology, microbiology, immunology, physiology, histology, anatomy or pathology. Documented skills in English corresponding to level 6/B. Intended learning outcomes The student will learn and understand the basis of quantitative genetic techniques, in particular how they pertain to the identification of genes underlying complex traits and diseases. Knowledge and understanding On completion of the course, the student shall be able to: Describe and understand statistical quantitative genetic techniques and how they apply to complex traits. Explain the statistical basis of quantitative traits. Explain and distinguish between linkage and linkage disequilibrium and their uses. Analyse the genetic architecture of different behavioral and disease-related traits. Analyse and understand the theory and steps required to identify the genetic components of a quantitative trait. -

Environment-Sensitive Epigenetics and the Heritability of Complex Diseases
INVESTIGATION Environment-Sensitive Epigenetics and the Heritability of Complex Diseases Robert E. Furrow,*,1 Freddy B. Christiansen,† and Marcus W. Feldman* *Department of Biology, Stanford University, Stanford, California 94305, and †Department of Bioscience and Bioinformatics Research Center, University of Aarhus, DK-8000 Aarhus C, Denmark ABSTRACT Genome-wide association studies have thus far failed to explain the observed heritability of complex human diseases. This is referred to as the “missing heritability” problem. However, these analyses have usually neglected to consider a role for epigenetic variation, which has been associated with many human diseases. We extend models of epigenetic inheritance to investigate whether environment-sensitive epigenetic modifications of DNA might explain observed patterns of familial aggregation. We find that variation in epigenetic state and environmental state can result in highly heritable phenotypes through a combination of epigenetic and environmental inheritance. These two inheritance processes together can produce familial covariances significantly higher than those predicted by models of purely epigenetic inheritance and similar to those expected from genetic effects. The results suggest that epigenetic variation, inherited both directly and through shared environmental effects, may make a key contribution to the missing heritability. HE challenges of identifying the common or rare genes a change in DNA sequence. Such modifications include Tthat contribute to the transmission of heritable human methylation of cytosine nucleotides at CpG sites and histone diseases and other complex phenotypes have been discussed protein modification. Such epigenetic modifications may be for some time (Moran 1973; Layzer 1974; Feldman and transmissible across generations or arise de novo each gen- Lewontin 1975; Kamin and Goldberger 2002). -

The Genetics of Tobacco Use: Methods, Findings and Policy Implications W Hall, P Madden, M Lynskey
119 Tob Control: first published as 10.1136/tc.11.2.119 on 1 June 2002. Downloaded from REVIEW The genetics of tobacco use: methods, findings and policy implications W Hall, P Madden, M Lynskey ............................................................................................................................. Tobacco Control 2002;11:119–124 Research on the genetics of smoking has increased our Specific study designs—adoption, twin, and link- understanding of nicotine dependence, and it is likely to age designs—are needed to separate the effects of genes from those of the environment. illuminate the mechanisms by which cigarette smoking adversely effects the health of smokers. Given recent Adoption studies advances in molecular biology, including the completion In the adoption design we compare the similarity in smoking between adoptive children and their of the draft sequence of the human genome, interest has biological parents with the similarity between now turned to identifying gene markers that predict a adopted children and their adoptive parents or heightened risk of using tobacco and developing between adoptive sibling pairs and biological sib- ling pairs. If smoking is largely or wholly geneti- nicotine dependence cally determined, then there should be greater .......................................................................... similarity between children and their biological parents and siblings than between adopted hanks to RA Fisher, genetics has been linked children and their adoptive parents or siblings. to scepticism about a causal relation between One early adoption study found an association cigarette smoking and lung cancer.12 Fisher3 between the smoking of foster children and their T biological siblings but not their adoptive argued that the association between smoking and 5 6 lung cancer was explained by shared genes that siblings. -

Epigenome-Wide Study Identified Methylation Sites Associated With
nutrients Article Epigenome-Wide Study Identified Methylation Sites Associated with the Risk of Obesity Majid Nikpay 1,*, Sepehr Ravati 2, Robert Dent 3 and Ruth McPherson 1,4,* 1 Ruddy Canadian Cardiovascular Genetics Centre, University of Ottawa Heart Institute, 40 Ruskin St–H4208, Ottawa, ON K1Y 4W7, Canada 2 Plastenor Technologies Company, Montreal, QC H2P 2G4, Canada; [email protected] 3 Department of Medicine, Division of Endocrinology, University of Ottawa, the Ottawa Hospital, Ottawa, ON K1Y 4E9, Canada; [email protected] 4 Atherogenomics Laboratory, University of Ottawa Heart Institute, Ottawa, ON K1Y 4W7, Canada * Correspondence: [email protected] (M.N.); [email protected] (R.M.) Abstract: Here, we performed a genome-wide search for methylation sites that contribute to the risk of obesity. We integrated methylation quantitative trait locus (mQTL) data with BMI GWAS information through a SNP-based multiomics approach to identify genomic regions where mQTLs for a methylation site co-localize with obesity risk SNPs. We then tested whether the identified site contributed to BMI through Mendelian randomization. We identified multiple methylation sites causally contributing to the risk of obesity. We validated these findings through a replication stage. By integrating expression quantitative trait locus (eQTL) data, we noted that lower methylation at cg21178254 site upstream of CCNL1 contributes to obesity by increasing the expression of this gene. Higher methylation at cg02814054 increases the risk of obesity by lowering the expression of MAST3, whereas lower methylation at cg06028605 contributes to obesity by decreasing the expression of SLC5A11. Finally, we noted that rare variants within 2p23.3 impact obesity by making the cg01884057 Citation: Nikpay, M.; Ravati, S.; Dent, R.; McPherson, R.