Identification of Nephrogenic Therapeutic Biomarkers of Wilms Tumor Using Machine Learning
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Upregulation of Peroxisome Proliferator-Activated Receptor-Α And
Upregulation of peroxisome proliferator-activated receptor-α and the lipid metabolism pathway promotes carcinogenesis of ampullary cancer Chih-Yang Wang, Ying-Jui Chao, Yi-Ling Chen, Tzu-Wen Wang, Nam Nhut Phan, Hui-Ping Hsu, Yan-Shen Shan, Ming-Derg Lai 1 Supplementary Table 1. Demographics and clinical outcomes of five patients with ampullary cancer Time of Tumor Time to Age Differentia survival/ Sex Staging size Morphology Recurrence recurrence Condition (years) tion expired (cm) (months) (months) T2N0, 51 F 211 Polypoid Unknown No -- Survived 193 stage Ib T2N0, 2.41.5 58 F Mixed Good Yes 14 Expired 17 stage Ib 0.6 T3N0, 4.53.5 68 M Polypoid Good No -- Survived 162 stage IIA 1.2 T3N0, 66 M 110.8 Ulcerative Good Yes 64 Expired 227 stage IIA T3N0, 60 M 21.81 Mixed Moderate Yes 5.6 Expired 16.7 stage IIA 2 Supplementary Table 2. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of an ampullary cancer microarray using the Database for Annotation, Visualization and Integrated Discovery (DAVID). This table contains only pathways with p values that ranged 0.0001~0.05. KEGG Pathway p value Genes Pentose and 1.50E-04 UGT1A6, CRYL1, UGT1A8, AKR1B1, UGT2B11, UGT2A3, glucuronate UGT2B10, UGT2B7, XYLB interconversions Drug metabolism 1.63E-04 CYP3A4, XDH, UGT1A6, CYP3A5, CES2, CYP3A7, UGT1A8, NAT2, UGT2B11, DPYD, UGT2A3, UGT2B10, UGT2B7 Maturity-onset 2.43E-04 HNF1A, HNF4A, SLC2A2, PKLR, NEUROD1, HNF4G, diabetes of the PDX1, NR5A2, NKX2-2 young Starch and sucrose 6.03E-04 GBA3, UGT1A6, G6PC, UGT1A8, ENPP3, MGAM, SI, metabolism -

Hexokinase 2 Is Targetable for HK1 Negative, HK2 Positive Tumors from a Wide Variety of Tissues of Origin
Journal of Nuclear Medicine, published on August 30, 2018 as doi:10.2967/jnumed.118.212365 Hexokinase 2 Is Targetable for HK1 Negative, HK2 Positive Tumors from a Wide Variety of Tissues of Origin Shili Xu1, Arthur Catapang1, Hanna M. Doh1, Nicholas A. Bayley1, Jason T. Lee1,2,3, Daniel Braas1,4, Thomas G. Graeber1,2,3,4, Harvey R. Herschman1,2,3,5,6* 1Department of Molecular and Medical Pharmacology 2Crump Institute for Molecular Imaging 3Jonsson Comprehensive Cancer Center 4UCLA Metabolomics Center 5Department of Biological Chemistry 6Molecular Biology Institute David Geffen School of Medicine, University of California Los Angeles, Los Angeles, California 90095, USA *Correspondence: Harvey R. Herschman. 341 Boyer Hall, UCLA, 611 Charles E. Young Drive East, Los Angeles CA, 90095. Phone: 310-825-8735. Fax: 310-825-1447. Email: [email protected] First author: Shili Xu, postdoctoral fellow. 329 Boyer Hall, UCLA, 611 Charles E. Young Drive East, Los Angeles CA, 90095. Phone: 310-825-8745. Email: [email protected] Running Title: HK2 Is Targetable for HK1-HK2+ Tumors Financial Disclosure: No potential conflicts of interest relevant to this article exist. Word count: 4997 1 ABSTRACT Although absent in most adult tissues, hexokinase 2 (HK2) is expressed in a majority of tumors and contributes to increased glucose consumption and to in vivo tumor 18F-FDG PET signaling. Methods: Both HK2 knockdown and knockout approaches were used to investigate the role of HK2 in cancer cell proliferation, in vivo xenograft tumor progression and 18F-FDG tumor accumulation. BioProfiler analysis monitored cell culture glucose consumption and lactate production; 18F-FDG PET/CT monitored in vivo tumor glucose accumulation. -

Gene Expression Signature for Biliary Atresia and a Role for Interleukin-8
SUPPLEMENTARY “PATIENTS AND METHODS” “Gene expression signature for biliary atresia and a role for Interleukin-8 in pathogenesis of experimental disease” Bessho K, et al. PATIENTS AND METHODS Patients. Liver biopsies, serum samples and clinical data were obtained from infants with cholestasis enrolled into a prospective study (ClinicalTrials.gov Identifier: NCT00061828) of the NIDDK-funded Childhood Liver Disease Research and Education Network (www.childrennetwork.org) or from infants evaluated at Cincinnati Children’s Hospital Medical Center. For subjects with biliary atresia (BA), liver biopsies were obtained from 64 infants during the preoperative workup or at the time of intraoperative cholangiogram, with ages ranging from 22-169 days after birth (Supplementary Table 7). For subjects with intrahepatic cholestasis (serving as diseased controls, and referred to as non- BA), liver biopsy samples were obtained percutaneously or intraoperatively from 14 infants at the time of diagnostic evaluation, with ages ranging from 19-189 days. Their diagnosis were Alagille syndrome (N=1), multidrug resistance protein-3 deficiency (N=2), alpha-1-antitrypsin deficiency (N=2) and cholestasis with unknown etiology (N=9) (Supplementary Table 7). Representative liver biopsy photomicrographs are shown in Supplementary Figure 6A-D. A third group of normal controls (NC) consisted of liver biopsy samples obtained from 7 deceased-donor children aged 22-42 months as described previously (1). This group serves as a reference cohort, with the median levels of gene expression used to normalize gene expression across all patients in the BA and non-BA groups. This greatly facilitates the visual identification of key differences in gene expression levels between BA and non-BA groups. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Downloaded from Here
bioRxiv preprint doi: https://doi.org/10.1101/017566; this version posted November 19, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Testing for ancient selection using cross-population allele 2 frequency differentiation 1;∗ 3 Fernando Racimo 4 1 Department of Integrative Biology, University of California, Berkeley, CA, USA 5 ∗ E-mail: [email protected] 6 1 Abstract 7 A powerful way to detect selection in a population is by modeling local allele frequency changes in a 8 particular region of the genome under scenarios of selection and neutrality, and finding which model is 9 most compatible with the data. Chen et al. [2010] developed a composite likelihood method called XP- 10 CLR that uses an outgroup population to detect departures from neutrality which could be compatible 11 with hard or soft sweeps, at linked sites near a beneficial allele. However, this method is most sensitive 12 to recent selection and may miss selective events that happened a long time ago. To overcome this, 13 we developed an extension of XP-CLR that jointly models the behavior of a selected allele in a three- 14 population tree. Our method - called 3P-CLR - outperforms XP-CLR when testing for selection that 15 occurred before two populations split from each other, and can distinguish between those events and 16 events that occurred specifically in each of the populations after the split. -

Current Status on Genome–Metabolome-Wide Associations: an Opportunity in Nutrition Research
Genes Nutr (2013) 8:19–27 DOI 10.1007/s12263-012-0313-7 REVIEW Current status on genome–metabolome-wide associations: an opportunity in nutrition research Ivan Montoliu • Ulrich Genick • Mirko Ledda • Sebastiano Collino • Franc¸ois-Pierre Martin • Johannes le Coutre • Serge Rezzi Received: 15 February 2012 / Accepted: 2 August 2012 / Published online: 16 October 2012 Ó Springer-Verlag 2012 Abstract Genome-wide association studies (GWASs) impact on the homeostatic concentrations of specific have become a very important tool to address the genetic metabolites. A particularly interesting aspect of this work origin of phenotypic variability, in particular associated takes into account interactions of environment and lifestyle with diseases. Nevertheless, these types of studies provide with the genome and how this interaction translates into limited information about disease etiology and the molec- changes in the metabolome. For instance, the role of PY- ular mechanisms involved. Recently, the incorporation of ROXD2 in trimethylamine metabolism points to an inter- metabolomics into the analysis has offered novel oppor- action between host and microbiome genomes (host/ tunities for a better understanding of disease-related met- microbiota). Often, these findings reveal metabolic dere- abolic deregulation. The pattern emerging from this work is gulations, which could eventually be tuned with a nutri- that gene-driven changes in metabolism are prevalent and tional intervention. Here we review the development of that common genetic variations can have a profound gene–metabolism association studies from a single-gene/ single-metabolite to a genome-wide/metabolome-wide approach and highlight the conceptual changes associated Ivan Montoliu and Ulrich Genick contributed equally to this work. -

NAT8 Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap4957c
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 NAT8 Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP4957c Specification NAT8 Antibody (Center) - Product Information Application WB, IHC-P,E Primary Accession Q9UHE5 Other Accession Q9UHF3 Reactivity Human Host Rabbit Clonality Polyclonal Isotype Rabbit Ig Antigen Region 110-138 NAT8 Antibody (Center) - Additional Information Gene ID 9027 Western blot analysis of NAT8 Antibody Other Names (Center) (Cat. #AP4957c) in ZR-75-1 cell line N-acetyltransferase 8, 231-, lysates (35ug/lane). NAT8 (arrow) was Acetyltransferase 2, ATase2, Camello-like detected using the purified Pab. protein 1, Cysteinyl-conjugate N-acetyltransferase, CCNAT, NAT8 (<a href ="http://www.genenames.org/cgi-bin/gene_ symbol_report?hgnc_id=18069" target="_blank">HGNC:18069</a>) Target/Specificity This NAT8 antibody is generated from rabbits immunized with a KLH conjugated synthetic peptide between 110-138 amino acids from the Central region of human NAT8. Dilution WB~~1:1000 IHC-P~~1:50~100 NAT8 Antibody (Center) (Cat. #AP4957c) IHC analysis in formalin fixed and paraffin Format embedded mouse kidney tissue followed by Purified polyclonal antibody supplied in PBS peroxidase conjugation of the secondary with 0.09% (W/V) sodium azide. This antibody and DAB staining. This data antibody is purified through a protein A demonstrates the use of the NAT8 Antibody column, followed by peptide affinity (Center) for immunohistochemistry. Clinical purification. relevance has not been evaluated. Storage Maintain refrigerated at 2-8°C for up to 2 NAT8 Antibody (Center) - Background weeks. For long term storage store at -20°C in small aliquots to prevent freeze-thaw This protein, isolated using the differential cycles. -

Product Datasheet NAT8 Overexpression
Product Datasheet NAT8 Overexpression Lysate NBL1-13491 Unit Size: 0.1 mg Store at -80C. Avoid freeze-thaw cycles. Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NBL1-13491 Updated 3/17/2020 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NBL1-13491 Page 1 of 2 v.20.1 Updated 3/17/2020 NBL1-13491 NAT8 Overexpression Lysate Product Information Unit Size 0.1 mg Concentration The exact concentration of the protein of interest cannot be determined for overexpression lysates. Please contact technical support for more information. Storage Store at -80C. Avoid freeze-thaw cycles. Buffer RIPA buffer Target Molecular Weight 25.4 kDa Product Description Description Transient overexpression lysate of N-acetyltransferase 8 (GCN5-related, putative) (NAT8) The lysate was created in HEK293T cells, using Plasmid ID RC203157 and based on accession number NM_003960. The protein contains a C-MYC/DDK Tag. Gene ID 9027 Gene Symbol NAT8 Species Human Notes HEK293T cells in 10-cm dishes were transiently transfected with a non-lipid polymer transfection reagent specially designed and manufactured for large volume DNA transfection. Transfected cells were cultured for 48hrs before collection. The cells were lysed in modified RIPA buffer (25mM Tris-HCl pH7.6, 150mM NaCl, 1% NP-40, 1mM EDTA, 1xProteinase inhibitor cocktail mix, 1mM PMSF and 1mM Na3VO4, and then centrifuged to clarify the lysate. Protein concentration was measured by BCA protein assay kit.This product is manufactured by and sold under license from OriGene Technologies and its use is limited solely for research purposes. -

Supplementary File 1
Supplementary Figure 1. Read coverage of the GI.1 and GI.2 genomes. The left panel shows viral genome coverage in rabbits infected with GI.1 and the right with GI.2. Red lines indicate adults, blue lines indicate kittens, and the highest coverage peak for each sample is annotated in grey text. Only samples with more than 20 reads mapping to the viral genomes are shown (see Table 1). Supplementary Figure 2. Significantly differentially expressed genes in infected rabbits compared to controls. Kittens infected with GI.2 showed a greater transcriptional response compared to the other treatment groups, evidenced by more differentially expressed genes. In particular, more genes were down- regulated and changes were clearly detectable by 12 hours post-infection. FDR is “false discovery rate” and FC is “fold change”. Supplementary Figure 3. Expression of genes in the “pink” co-expression module (a) and change in selected genes over time (b). Genes in this co- expression cluster tended to be up-regulated in control kittens, further up-regulated in GI.1-infected kittens but down-regulated in GI.2-infected kittens, suggesting a role in the resistance of kittens to GI.1. Gene expression was normalised using reads per kilobase per million (RPKM). In (b) the header in each plot gives the Ensembl gene accession followed by the gene symbol. Supplementary Table S1. The top 10 most differentially expressed genes in GI.2‐infected rabbits. Ensembl gene # Annotation Log fold FDR* change Kittens after 12 hours ENSOCUG00000024623 N/A ‐6.2 1.04E‐15 ENSOCUG00000002051 -

Apoptotic Cells Inflammasome Activity During the Uptake of Macrophage
Downloaded from http://www.jimmunol.org/ by guest on October 2, 2021 is online at: average * The Journal of Immunology published online 20 April 2012 from submission to initial decision 4 weeks from acceptance to publication http://www.jimmunol.org/content/early/2012/04/20/jimmun ol.1103760 Complement Protein C1q Directs Macrophage Polarization and Limits Inflammasome Activity during the Uptake of Apoptotic Cells Marie E. Benoit, Elizabeth V. Clarke, Pedro Morgado, Deborah A. Fraser and Andrea J. Tenner J Immunol Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://www.jimmunol.org/content/suppl/2012/04/20/jimmunol.110376 0.DC1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of October 2, 2021. Published April 20, 2012, doi:10.4049/jimmunol.1103760 The Journal of Immunology Complement Protein C1q Directs Macrophage Polarization and Limits Inflammasome Activity during the Uptake of Apoptotic Cells Marie E. Benoit, Elizabeth V. Clarke, Pedro Morgado, Deborah A. Fraser, and Andrea J. Tenner Deficiency in C1q, the recognition component of the classical complement cascade and a pattern recognition receptor involved in apoptotic cell clearance, leads to lupus-like autoimmune diseases characterized by auto-antibodies to self proteins and aberrant innate immune cell activation likely due to impaired clearance of apoptotic cells. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -
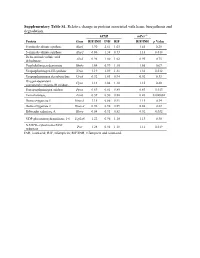
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator-