Studying How Components of the Action Potential Waveform
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Long QT Syndrome: from Channels to Cardiac Arrhythmias
Long QT syndrome: from channels to cardiac arrhythmias Arthur J. Moss, Robert S. Kass J Clin Invest. 2005;115(8):2018-2024. https://doi.org/10.1172/JCI25537. Review Series Long QT syndrome, a rare genetic disorder associated with life-threatening arrhythmias, has provided a wealth of information about fundamental mechanisms underlying human cardiac electrophysiology that has come about because of truly collaborative interactions between clinical and basic scientists. Our understanding of the mechanisms that control the critical plateau and repolarization phases of the human ventricular action potential has been raised to new levels through these studies, which have clarified the manner in which both potassium and sodium channels regulate this critical period of electrical activity. Find the latest version: https://jci.me/25537/pdf Review series Long QT syndrome: from channels to cardiac arrhythmias Arthur J. Moss1 and Robert S. Kass2 1Heart Research Follow-up Program, Department of Medicine, University of Rochester School of Medicine and Dentistry, Rochester, New York, USA. 2Department of Pharmacology, Columbia University Medical Center, New York, New York, USA. Long QT syndrome, a rare genetic disorder associated with life-threatening arrhythmias, has provided a wealth of information about fundamental mechanisms underlying human cardiac electrophysiology that has come about because of truly collaborative interactions between clinical and basic scientists. Our understanding of the mecha- nisms that control the critical plateau and repolarization phases of the human ventricular action potential has been raised to new levels through these studies, which have clarified the manner in which both potassium and sodium channels regulate this critical period of electrical activity. -

Neurotransmitter Actions
Central University of South Bihar Panchanpur, Gaya, India E-Learning Resources Department of Biotechnology NB: These materials are taken/borrowed/modified/compiled from various resources like research articles and freely available internet websites, and are meant to be used solely for the teaching purpose in a public university, and for serving the needs of specified educational programmes. Dr. Jawaid Ahsan Assistant Professor Department of Biotechnology Central University of South Bihar (CUSB) Course Code: MSBTN2003E04 Course Name: Neuroscience Neurotransmitter Actions • Excitatory Action: – A neurotransmitter that puts a neuron closer to an action potential (facilitation) or causes an action potential • Inhibitory Action: – A neurotransmitter that moves a neuron further away from an action potential • Response of neuron: – Responds according to the sum of all the neurotransmitters received at one time Neurotransmitters • Acetylcholine • Monoamines – modified amino acids • Amino acids • Neuropeptides- short chains of amino acids • Depression: – Caused by the imbalances of neurotransmitters • Many drugs imitate neurotransmitters – Ex: Prozac, zoloft, alcohol, drugs, tobacco Release of Neurotransmitters • When an action potential reaches the end of an axon, Ca+ channels in the neuron open • Causes Ca+ to rush in – Cause the synaptic vesicles to fuse with the cell membrane – Release the neurotransmitters into the synaptic cleft • After binding, neurotransmitters will either: – Be destroyed in the synaptic cleft OR – Taken back in to surrounding neurons (reuptake) Excitable cells: Definition: Refers to the ability of some cells to be electrically excited resulting in the generation of action potentials. Neurons, muscle cells (skeletal, cardiac, and smooth), and some endocrine cells (e.g., insulin- releasing pancreatic β cells) are excitable cells. -

1 the New Definition of Early Repolarisation Peter W. Macfarlane
1 The New Definition of Early Repolarisation Peter W. Macfarlane Institute of Cardiovascular and Medical Sciences University of Glasgow Address for Correspondence: Electrocardiology Group Level 1, New Lister Building Royal Infirmary, Glasgow G31 2ER Scotland, UK. e-mail: [email protected] 2 Introduction There has been an increasing interest in the electrocardiographic pattern of early repolarization in recent years following the publication of a paper by Haissaguerre and colleagues [1] in 2008, which linked their definition of the pattern to sudden cardiac arrest due to idiopathic ventricular fibrillation. However, earlier and later publications [2-17] by other groups on the topic of early repolarisation seemed to exhibit disagreement over what constituted the ECG pattern of early repolarisation suggesting a degree of confusion following the 2008 publication [1]. In 1947, Myers et al [2] reported that, among other features, an “elevation of the RS-T take off of 0.5 - 2.0mm may be considered as a normal variant” provided that the ST segment was concave upwards and followed by a tall T wave. In 1951, Goldman also reported on RS-T segment elevation in V3-V6 as a normal variant [3]. The original description of what for many years was regarded as the early repolarization pattern can probably be traced back to an early paper by Wasserburger and Alt [4] dealing with normal RS-T elevation in the relatively newly established full 12 lead ECG. The authors pointed out that the ST elevation mostly arose from a distinct notch on the downslope of the R wave but was occasionally associated with a “well-defined slur”. -

The Short QT Syndrome
The Short QT Syndrome A note from the SADS Foundation References We provide this information with the hope that informing 1. Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman A Guide for Patients BR, Bjerregaard P. Idiopathic short QT interval: a new clinical physicians and other health care providers, as well as the public, and Health Care will encourage early and correct diagnosis and proper therapy syndrome? Cardiology 2000;94:99 –102. for congenital short QT syndrome (SQTS), resulting in the 2. Giustetto C, Di Monte F. Wolpert C, Borggrefe M, Schimpf R, Providers reduction and ultimately elimination of sudden cardiac arrest Sbragia P, Leone G, Maury P, Anttonen O, Haissaguerre M, Gaita F. (SCA) and sudden cardiac death (SCD). Short QTsyndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006;27:2440 –2447. 3. Gaita F, Giustetto C, Bianchi F, Wolptert C, Schimpf R, Riccardi R, Grossi S, Richiardi E, Borggrefe M. Short QT syndrome: a familial What do Patients and Parents Need to Know cause of sudden death. Circulation 2003;108:965–970. about SQTS? 4. Dhutia H, Malhotra A, Parpia S, et al. The Prevalence and • The warning signs and symptoms of SQTS. significance of a short QT interval in 18 825 low-risk individuals • Who to see for proper testing. including athletes. Br J Sports Med 2015;0:1-6 • How to protect their children and themselves. • How to expand their family pedigree and contact other family members who may be at risk What do Physicians need to know? • When to consider SQTS as a possible diagnosis. -
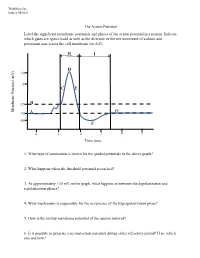
The Action Potential Label the Significant Membrane Potentials and Phases of the Action Potential in a Neuron
Worksheet by Andrey Michel The Action Potential Label the significant membrane potentials and phases of the action potential in a neuron. Indicate which gates are open/closed as well as the direction of the net movement of sodium and potassium ions across the cell membrane for A-G. H I D +30 0 C E -55 B Membrane Potential Membrane (mV) G -70 A -80 F 0 1 2 3 4 5 Time (ms) 1. What type of summation is shown for the graded potentials in the above graph? 2. What happens when the threshold potential is reached? 3. At approximately +30 mV on the graph, what happens in between the depolarization and repolarization phases? 4. What mechanism is responsible for the occurrence of the hyperpolarization phase? 5. How is the resting membrane potential of the neuron restored? 6. Is it possible to generate a second action potential during either refractory period? If so, which one and how? Place the following events in chronological order from 1-8: Na+ enters the cell, and depolarization occurs to approximately +30 mV. The voltage across the cell membrane is -70 mV, the resting membrane potential. Upon reaching the peak of the action potential, the VG Na+ channels are inactivated by the closing of their inactivation gate and the activation gate of each VG K+ channel opens. VG K+ channels close by the closing of their activation gate, and the resting membrane potential is gradually restored. An excitatory post-synaptic potential depolarizes the membrane to threshold and the activation gate of VG Na+ channels open. -

QT Interval Variations and Mortality Risk: Is There Any Relationship?
Education 255 QT interval variations and mortality risk: Is there any relationship? 1 Ciprian Rezuş, Victor Dan Moga , Anca Ouatu, Mariana Floria III Medical Clinic of “Sf. Spiridon” University Hospital, and “Grigore T. Popa” University of Medicine and Pharmacy; Iaşi-Romania 1Cardiology Clinic of Emergency Hospital and Victor Babes University of Medicine and Pharmacy; Timisoara-Romania ABSTRACT Abnormal prolongation and shortening of the electrocardiographic QT interval duration, which occur in the hereditary forms of long and short QT syndromes, are associated with an increased risk of ventricular arrhythmias and sudden cardiac death. Even within the normal range, these altered durations are associated with an increased mortality risk in the general population. While extreme prolongation or reduction of the QT interval predisposes patients to malignant ventricular arrhythmias and sudden cardiac death, the precise dose-response relationship between the QT interval and cardiovascular disease mortality is still unknown. This paper describes the need for more standardized methods for measur- ing and reporting the QT interval and the need for more precise assessments of the risk associated with QT interval variation. (Anatol J Cardiol 2015; 15: 255-8) Keywords: QT interval, QTc, mortality, risk, ventricular arrhythmias Introduction values for the QTc range from 350 to 450 ms for adult men and from 360 to 460 ms for adult women; however, 10%-20% of oth- Prolongation of the QT interval is associated with early after- erwise healthy persons may have QTc values outside this range. depolarizations. Early afterdepolarizations of sufficient ampli- Marked prolongations in the QT interval may be caused by tude can generate premature action potentials that lead to car- genetic disorders (e.g., long QT syndrome), pharmacological diac arrhythmias, which can progress to ventricular fibrillation agents (e.g., antiarrhythmics, antipsychotics, and antibiotics), and sudden cardiac death. -

Silent and Malignant Early Repolarization Syndrome Mimicking Hyper-Acute ST Elevation Myocardial Infarction
Acta Cardiol Sin 2016;32:506-510 Case Report doi: 10.6515/ACS20151012A Silent and Malignant Early Repolarization Syndrome Mimicking Hyper-Acute ST Elevation Myocardial Infarction Weng-Chio Tam,1,2 Ming-Hsiung Hsieh,1,2 Yung-Kuo Lin1,2 and Jong-Shiuan Yeh1,2 A 55-year-old male with underlying type 2 diabetes mellitus and hypertension presented at our emergency department with ventricular fibrillation-related cardiac arrest. Hyper-acute ST elevation myocardial infarction was the preliminary diagnosis by 12-lead electrocardiography, which simultaneously showed J point ST elevation and tall T waves. However, the echocardiography showed concentric left ventricle hypertrophy and preserved left ventricular systolic function with no regional wall motion abnormalities, and coronary angiography did not show anycriticalcoronaryarterylesion.Malignantearlyrepolarization syndrome was diagnosed, and an implantable cardioverter defibrillator was implanted. Early repolarization syndrome is associated with J point elevation, and more involved leads and an increased J point elevation amplitude can increase the risk of arrhythmogenicity. In summary, we report a case with asymptomatic type 3 early repolarization syndrome-induced idiopathic ventricular fibrillation mimicking hyper-acute ST elevation myocardial infarction. Key Words: Early repolarization · Idiopathic ventricular fibrillation · J wave syndrome INTRODUCTION the first case report of its type in Taiwan. For an asymp- tomatic early repolarization pattern, distinguishing ma- Herein,wehavepresentedthecaseofapatient -

ATP-Sensitive Potassium Channels: a Review of Their Cardioprotective Pharmacology
J Mol Cell Cardiol 32, 677±695 (2000) doi:10.1006/jmcc.2000.1111, available online at http://www.idealibrary.com on ATP-Sensitive Potassium Channels: A Review of their Cardioprotective Pharmacology Gary J. Grover1 and Keith D. Garlid2 1 Cardiovascular and Metabolic Diseases Drug Discovery, Bristol-Myers Squibb Pharmaceutical Research Institute, PO Box 4000, Princeton, N.J. 08543-4000, USA; 2 Department of Biochemistry, and Molecular Biology, Oregon Graduate Institute of Science and Technology, 200000 NW Walker Road, Beaverton, Oregon 97006-8291 (Received 8 September 1999, accepted in revised form 27 January 2000) G. J. G K. A. G. ATP-Sensitive Potassium Channels: A Review of their Cardioprotective Phar- macology. Journal of Molecular and Cellular Cardiology (2000) 32, 677±696. ATP-sensitive potassium channels (KATP) have been thought to be a mediator of cardioprotection for the last ten years. Signi®cant progress has been made in learning the pharmacology of this channel as well as its molecular regulation with regard to cardioprotection. KATP openers as a class protect ischemic/reperfused myocardium and appear to do so by conservation of energy. The reduced rate of ATP hydrolysis during ischemia exerted by these openers is not due to a cardioplegic effect and is independent of action potential shortening. Compounds have been synthesized which retain the cardioprotective effects of ®rst generation KATP openers, but are devoid of vasodilator and cardiac sarcolemmal potassium outward currents. These results suggest receptor or channel subtypes. Recent pharmacologic and molecular biology studies suggest the activation of mitochondrial KATP as the relevant cardioprotective site. Implications of these results for future drug discovery and preconditioning are discussed. -

Kv2 Channel Regulation of Action Potential Repolarization and Firing Patterns in Superior Cervical Ganglion Neurons and Hippocampal CA1 Pyramidal Neurons
The Journal of Neuroscience, April 2, 2014 • 34(14):4991–5002 • 4991 Cellular/Molecular Kv2 Channel Regulation of Action Potential Repolarization and Firing Patterns in Superior Cervical Ganglion Neurons and Hippocampal CA1 Pyramidal Neurons Pin W. Liu and Bruce P. Bean Department of Neurobiology, Harvard Medical School, Boston, Massachusetts 02115 Kv2 family “delayed-rectifier” potassium channels are widely expressed in mammalian neurons. Kv2 channels activate relatively slowly and their contribution to action potential repolarization under physiological conditions has been unclear. We explored the function of Kv2 channels using a Kv2-selective blocker, Guangxitoxin-1E (GxTX-1E). Using acutely isolated neurons, mixed voltage-clamp and current-clamp experiments were done at 37°C to study the physiological kinetics of channel gating and action potentials. In both rat superior cervical ganglion (SCG) neurons and mouse hippocampal CA1 pyramidal neurons, 100 nM GxTX-1E produced near-saturating block of a component of current typically constituting ϳ60–80% of the total delayed-rectifier current. GxTX-1E also reduced A-type potassium current (IA ), but much more weakly. In SCG neurons, 100 nM GxTX-1E broadened spikes and voltage clamp experiments using action potential waveforms showed that Kv2 channels carry ϳ55% of the total outward current during action potential repolarization despite activating relatively late in the spike. In CA1 neurons, 100 nM GxTX-1E broadened spikes evoked from Ϫ70 mV, but not Ϫ80 mV, likely reflecting a greater role of Kv2 when other potassium channels were partially inactivated at Ϫ70 mV. In both CA1 and SCG neurons, inhibition of Kv2 channels produced dramatic depolarization of interspike voltages during repetitive firing. -

The Repolarization Phase of the Cardiac Ventricular Action Potential: a Time-Dependent System of Membrane Conductances
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector THE REPOLARIZATION PHASE OF THE CARDIAC VENTRICULAR ACTION POTENTIAL: A TIME-DEPENDENT SYSTEM OF MEMBRANE CONDUCTANCES E. A. JOHNSON and J. TILLE From the Department of Pharmacology, University of Sydney, Sydney, Australia. Dr. Johnson's present address is the Department of Physiology and Pharmacology, Duke University, Durham, North Carolina. Mr. Tille's present address is the De- partment of Physiology, Monash University, Clayton, Victoria, Australia ABSTRACT. A system for the generation of the repolarization phase of the ventricular action potential is described. The system is based on time-depend- ent changes in membrane conductance to sodium and potassium ions. How- ever, the changes in conductance during an action potential retain a degree of voltage dependence through the initial conditions which depend on previous depolarizations of the membrane. The equations describing the system were solved with an analog computer and various action potential forms are re- produced. The effects of hyperpolarizing and depolarizing current applied dur- ing an action potential are investigated. The changes in shape of an action potential after a change in the rate of stimulation show partial agreement with previous experimental findings. The applicability of time-dependent and voltage- dependent systems for the generation of the repolarization phase of the ventri- cular action potential is discussed. INTRODUCTION The success of the Hodgkin and Huxley equations (HH,1 1952) in describing the generation of the action potential in squid giant axon has led to attempts to use these equations in a more or less modified form for a description of the generation of the cardiac action potential. -

Molecular Studies of ATP-Sensitive Potassium Channels : Gating, Pathology, and Optogenetics Gina Catalina Reyes-Mejia
Molecular studies of ATP-sensitive potassium channels : gating, pathology, and optogenetics Gina Catalina Reyes-Mejia To cite this version: Gina Catalina Reyes-Mejia. Molecular studies of ATP-sensitive potassium channels : gating, pathol- ogy, and optogenetics. Biomolecules [q-bio.BM]. Université Grenoble Alpes, 2016. English. NNT : 2016GREAV012. tel-01497530 HAL Id: tel-01497530 https://tel.archives-ouvertes.fr/tel-01497530 Submitted on 28 Mar 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE LA COMMUNAUTE UNIVERSITE GRENOBLE ALPES Spécialité : Biologie Structurale et Nanobiologie Arrêté ministériel : 7 août 2006 Présentée par Gina Catalina REYES MEJIA Thèse dirigée par Dr. Michel VIVAUDOU préparée au sein du l’institut de Biologie Structurale dans l'École Doctorale Chimie et Science du Vivant Etudes moléculaires du canal potassique sensible à l'ATP:"gating", pathologie et optogénétique Thèse soutenue publiquement le 23 Septembre 2016 devant le jury composé de : Pr. Franck FIESCHI Président Dr. Thomas GRUTTER Rapporteur Dr. Gildas LOUSSOUARN Rapporteur Dr. Guillaume SANDOZ Examinateur Dr. Michel VIVAUDOU Directeur de thèse Université de Grenoble École Doctorale Chimie et Science du Vivant Thèse pour obtenir le grade de DOCTEUR EN SCIENCES DE L’UNIVERSITÉ DE GRENOBLE Specialité: Biologie Structurale et Nanobiologie Presented and defended in public by Gina Catalina REYES MEJIA On September 23th 2016 Molecular studies of ATP-sensitive potassium channels: Gating, pathology, and optogenetics Thesis director: Dr. -

HEREDITARY, CONGENITAL, FAMILIAL SHORT-QT SYNDROME Or “GUSSAK SYNDROME”
HEREDITARY, CONGENITAL, FAMILIAL SHORT-QT SYNDROME or “GUSSAK SYNDROME” Andrés Ricardo Pérez Riera MD1 1 Chief of Electro-Vectocardiology Sector of the Discipline of Cardiology, ABC Faculty of Medicine (FMABC), Foundation of ABC (FUABC) – Santo André – Sao Paulo – Brazil. E-mail: [email protected] Correspondence: Andrés Ricardo Pérez Riera, MD Rua Sebastião Afonso, 885 - Jd. Miriam Zip Code: 04417- 100 – Sao Paulo – Brazil Phone: (55 11) 5621-2390 Fax: (55 11) 5625-7278/ 5506-0398 E-mail: [email protected] Key words: Electrocardiography • Arrhythmia • Sudden death • Short QT syndrome • Gene mutation • 1 Abstract Hereditary, congenital or familial short QT syndrome is a clinical electrocardiographic, familial hereditary channelopathy, autosomal dominant or sporadic, genetically heterogeneous of the electrical system of the heart that consists of a constellation of signs and symptoms, as syncope, sudden death, palpitations and dizziness with high tendency to paroxysmal atrial fibrillation (AF) episodes, short QT interval on ECG (QTc ≤ 300 ms) that doesn't significantly change with heart rate, tall, narrow-based and peaked T waves that are reminiscent of the typical T waves in "desert tent" of hyperkalemia, structurally normal heart, short refractory periods in atrial and ventricular myocytes and inducible ventricular fibrillation to programmed electrical stimulation. A few affected families have been identified. There are sporadic cases. Familial short QT syndrome is a clinical and genetic counterpart, the long-QT syndrome. Both syndromes reflect the close, but opposite relationship of potassium channel dysfunction in the setting of normal repolarization. Chronology of the discovery Year 1993: The first data suggesting that not only too long but also too short a QT interval (<400 ms) may be associated with increased risk of sudden death (SD)1.