Catalytic Cracking of N-Hexadecane Using Carbon Nanostructures/Nano-Zeolite-Y Composite Catalyst
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

N-Hexadecane Fuel for a Phosphoric Acid Direct Hydrocarbon Fuel Cell
Hindawi Publishing Corporation Journal of Fuels Volume 2015, Article ID 748679, 9 pages http://dx.doi.org/10.1155/2015/748679 Research Article n-Hexadecane Fuel for a Phosphoric Acid Direct Hydrocarbon Fuel Cell Yuanchen Zhu,1,2 Travis Robinson,1 Amani Al-Othman,2,3 André Y. Tremblay,1 and Marten Ternan4 1 Chemical and Biological Engineering, University of Ottawa, 161 Louis Pasteur, Ottawa, ON, Canada K1N 6N5 2Catalysis Centre for Research and Innovation, University of Ottawa, 30 Marie Curie, Ottawa, ON, Canada K1N 6N5 3Chemical Engineering, American University of Sharjah, Sharjah, UAE 4EnPross Incorporated, 147 Banning Road, Ottawa, ON, Canada K2L 1C5 Correspondence should be addressed to Marten Ternan; [email protected] Received 8 January 2015; Accepted 17 March 2015 Academic Editor: Michele Gambino Copyright © 2015 Yuanchen Zhu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The objective of this work was to examine fuel cells as a possible alternative to the diesel fuel engines currently used inrailway locomotives, thereby decreasing air emissions from the railway transportation sector. We have investigated the performance of a phosphoric acid fuel cell (PAFC) reactor, with n-hexadecane, C16H34 (a model compound for diesel fuel, cetane number = 100). This is the first extensive study reported in the literature in which n-hexadecane is used directly as the fuel. Measurements were made to obtain both polarization curves and time-on-stream results. Because deactivation was observed hydrogen polarization curves were measured before and after n-hexadecane experiments, to determine the extent of deactivation of the membrane electrode assembly (MEA). -

Synthetic Turf Scientific Advisory Panel Meeting Materials
California Environmental Protection Agency Office of Environmental Health Hazard Assessment Synthetic Turf Study Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019 MEETING MATERIALS THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency Agenda Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019, 9:30 a.m. – 4:00 p.m. 1001 I Street, CalEPA Headquarters Building, Sacramento Byron Sher Auditorium The agenda for this meeting is given below. The order of items on the agenda is provided for general reference only. The order in which items are taken up by the Panel is subject to change. 1. Welcome and Opening Remarks 2. Synthetic Turf and Playground Studies Overview 4. Synthetic Turf Field Exposure Model Exposure Equations Exposure Parameters 3. Non-Targeted Chemical Analysis Volatile Organics on Synthetic Turf Fields Non-Polar Organics Constituents in Crumb Rubber Polar Organic Constituents in Crumb Rubber 5. Public Comments: For members of the public attending in-person: Comments will be limited to three minutes per commenter. For members of the public attending via the internet: Comments may be sent via email to [email protected]. Email comments will be read aloud, up to three minutes each, by staff of OEHHA during the public comment period, as time allows. 6. Further Panel Discussion and Closing Remarks 7. Wrap Up and Adjournment Agenda Synthetic Turf Advisory Panel Meeting May 31, 2019 THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency DRAFT for Discussion at May 2019 SAP Meeting. Table of Contents Synthetic Turf and Playground Studies Overview May 2019 Update ..... -
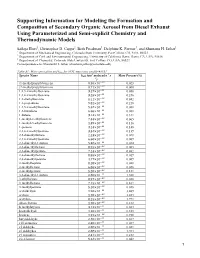
Supporting Information for Modeling the Formation and Composition Of
Supporting Information for Modeling the Formation and Composition of Secondary Organic Aerosol from Diesel Exhaust Using Parameterized and Semi-explicit Chemistry and Thermodynamic Models Sailaja Eluri1, Christopher D. Cappa2, Beth Friedman3, Delphine K. Farmer3, and Shantanu H. Jathar1 1 Department of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA, 80523 2 Department of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA, 95616 3 Department of Chemistry, Colorado State University, Fort Collins, CO, USA, 80523 Correspondence to: Shantanu H. Jathar ([email protected]) Table S1: Mass speciation and kOH for VOC emissions profile #3161 3 -1 - Species Name kOH (cm molecules s Mass Percent (%) 1) (1-methylpropyl) benzene 8.50×10'() 0.023 (2-methylpropyl) benzene 8.71×10'() 0.060 1,2,3-trimethylbenzene 3.27×10'(( 0.056 1,2,4-trimethylbenzene 3.25×10'(( 0.246 1,2-diethylbenzene 8.11×10'() 0.042 1,2-propadiene 9.82×10'() 0.218 1,3,5-trimethylbenzene 5.67×10'(( 0.088 1,3-butadiene 6.66×10'(( 0.088 1-butene 3.14×10'(( 0.311 1-methyl-2-ethylbenzene 7.44×10'() 0.065 1-methyl-3-ethylbenzene 1.39×10'(( 0.116 1-pentene 3.14×10'(( 0.148 2,2,4-trimethylpentane 3.34×10'() 0.139 2,2-dimethylbutane 2.23×10'() 0.028 2,3,4-trimethylpentane 6.60×10'() 0.009 2,3-dimethyl-1-butene 5.38×10'(( 0.014 2,3-dimethylhexane 8.55×10'() 0.005 2,3-dimethylpentane 7.14×10'() 0.032 2,4-dimethylhexane 8.55×10'() 0.019 2,4-dimethylpentane 4.77×10'() 0.009 2-methylheptane 8.28×10'() 0.028 2-methylhexane 6.86×10'() -

Safety Data Sheet
SAFETY DATA SHEET Flammable Liquid Mixture: Docosane / Dodecane / Eicosane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Heptane / N-Nonane / N-Octadecane / N- Octane / Octacosane / Tetracosane / Tetradecane Section 1. Identification GHS product identifier : Flammable Liquid Mixture: Docosane / Dodecane / Eicosane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Heptane / N-Nonane / N-Octadecane / N-Octane / Octacosane / Tetracosane / Tetradecane Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 019735 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 1 substance or mixture SKIN CORROSION/IRRITATION - Category 2 SERIOUS EYE DAMAGE/ EYE IRRITATION - Category 2A TOXIC TO REPRODUCTION (Fertility) - Category 2 TOXIC TO REPRODUCTION (Unborn child) - Category 2 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Respiratory tract irritation) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (REPEATED EXPOSURE) - Category 2 AQUATIC HAZARD (ACUTE) - Category 2 AQUATIC HAZARD (LONG-TERM) - Category 1 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable liquid and vapor. May form explosive mixtures in Air. Causes serious eye irritation. Causes skin irritation. May cause respiratory irritation. Suspected of damaging fertility or the unborn child. May cause drowsiness and dizziness. May cause damage to organs through prolonged or repeated exposure. Very toxic to aquatic life with long lasting effects. Precautionary statements General : Read label before use. -

The Strength in Numbers: Comprehensive Characterization of House Dust Using Complementary Mass Spectrometric Techniques
Analytical and Bioanalytical Chemistry https://doi.org/10.1007/s00216-019-01615-6 PAPER IN FOREFRONT The strength in numbers: comprehensive characterization of house dust using complementary mass spectrometric techniques Pawel Rostkowski1 & Peter Haglund2 & Reza Aalizadeh3 & Nikiforos Alygizakis 3,4 & Nikolaos Thomaidis 3 & Joaquin Beltran Arandes5 & Pernilla Bohlin Nizzetto1 & Petra Booij 6 & Hélène Budzinski7 & Pamela Brunswick8 & Adrian Covaci9 & Christine Gallampois2 & Sylvia Grosse10 & Ralph Hindle11 & Ildiko Ipolyi4 & Karl Jobst12 & Sarit L. Kaserzon13 & Pim Leonards14 & Francois Lestremau15 & Thomas Letzel10 & Jörgen Magnér16,17 & Hidenori Matsukami18 & Christoph Moschet19 & Peter Oswald 4 & Merle Plassmann20 & Jaroslav Slobodnik4 & Chun Yang21 Received: 6 November 2018 /Revised: 20 December 2018 /Accepted: 15 January 2019 # The Author(s) 2019 Abstract Untargeted analysis of a composite house dust sample has been performed as part of a collaborative effort to evaluate the progress in the field of suspect and nontarget screening and build an extensive database of organic indoor environment contaminants. Twenty- one participants reported results that were curated by the organizers of the collaborative trial. In total, nearly 2350 compounds were identified (18%) or tentatively identified (25% at confidence level 2 and 58% at confidence level 3), making the collaborative trial a success. However, a relatively small share (37%) of all compounds were reported by more than one participant, which shows that there is plenty of room for improvement in the field of suspect and nontarget screening. An even a smaller share (5%) of the total number of compounds were detected using both liquid chromatography–mass spectrometry (LC-MS) and gas chromatography– mass spectrometry (GC-MS). Thus, the two MS techniques are highly complementary. -

P1 What Is Petroleum?
P1 WHAT IS PETROLEUM? Petroleum Petroleum is a mixture of several different hydrocarbons; the most commonly found molecules are alkanes (linear or branched), cycloalkanes, aromatic hydrocarbons, or more complicated chemicals like asphaltenes. Each petroleum variety has a unique mix of molecules which defines its physical and chemical properties, like colour and viscosity. The alkanes, also known as paraffins, are saturated hydrocarbons with straight or branched chains which contain only carbon and hydrogen and have the general formula CnH2n+2. They generally have from 5 to 40 carbon atoms per molecule, although trace amounts Arial photograph of Sasolburg of shorter or longer molecules may be present in the mixture. Two representations of octane, a hydrocarbon The alkanes from pentane (C5H12) to octane (C8H18) are refined into petrol; the ones from nonane (C H ) found in petroleum 9 20 to hexadecane (C16H34) into diesel fuel and kerosene (primary component of many types of jet fuel); and the ones from hexadecane upwards into fuel oil and lubricating oil. At the heavier end of the range, paraffin wax is an alkane with approximately 25 carbon atoms, while asphalt has 35 carbon atoms or more. These are usually cracked (split) by modern refineries into more valuable products. Source: Wikimedia Commons The shortest molecules, those with four or fewer carbon A diagram of crude oil distillation atoms, are in a gaseous state at room temperature. as They are the petroleum gases. 20C Depending on demand and the cost of recovery, these gases are either flared off, sold as liquefied petroleum gas under pressure, or used to power the refinery's 10C own burners. -

Densities of the Binary Systems N-Hexane + N-Decane and N-Hexane + N-Hexadecane up to 60 Mpa and 463 K
Downloaded from orbit.dtu.dk on: Sep 25, 2021 Densities of the Binary Systems n-Hexane + n-Decane and n-Hexane + n-Hexadecane up to 60 MPa and 463 K Regueira , Teresa; Yan, Wei; Stenby, Erling H. Published in: Journal of Chemical and Engineering Data Link to article, DOI: 10.1021/acs.jced.5b00613 Publication date: 2015 Document Version Peer reviewed version Link back to DTU Orbit Citation (APA): Regueira , T., Yan, W., & Stenby, E. H. (2015). Densities of the Binary Systems n-Hexane + n-Decane and n- Hexane + n-Hexadecane up to 60 MPa and 463 K. Journal of Chemical and Engineering Data, 60(12), 3631- 3645. https://doi.org/10.1021/acs.jced.5b00613 General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Densities of the binary systems n-hexane + n-decane and n-hexane + n-hexadecane up to 60 MPa and 463 K Teresa Regueira, Wei Yan*, Erling H. -

Diesel) Engines Bioblendstocks with Potential for Decreased Emissions and Improved Operability
Co-Optimization of Fuels & Engines Top 13 Blendstocks Derived from Biomass for Mixing-Controlled Compression-Ignition (Diesel) Engines Bioblendstocks with Potential for Decreased Emissions and Improved Operability Co-Optimization of Fuels & Engines Top 13 MCCI Bioblendstocks About the Co-Optimization of Fuels & Engines Project This is one of a series of reports produced by the Co-Optimization of Fuels & Engines (Co-Optima) project, which is a U.S. Department of Energy (DOE)-sponsored multi-agency project that was initiated to accelerate the introduction of affordable, scalable, and sustainable biofuels and high-efficiency, low-emission vehicle engines. The simultaneous fuels and vehicles research and development is designed to deliver maximum energy savings, emissions reduction, and on-road performance. Co-Optima brings together two DOE Office of Energy Efficiency and Renewable Energy (EERE) research offices, nine national laboratories, and numerous industry and academic partners to make improvements to the types of fuels and engines found in most vehicles currently on the road and to develop revolutionary engine technologies for longer-term, higher-impact solutions. This first-of-its-kind project will provide industry with the scientific underpinnings required to move new biofuels and advanced engine systems to market faster while identifying and addressing barriers to commercialization. In addition to the EERE Vehicle Technologies and Bioenergy Technologies Offices, the Co- Optima project team included representatives from the National Renewable Energy Laboratory and Argonne, Idaho, Lawrence Berkeley, Lawrence Livermore, Los Alamos, Oak Ridge, Pacific Northwest, and Sandia National Laboratories. More detail on the project, as well as the full series of reports, can be found at www.energy.gov/fuel-engine-co-optimization. -

Plasma Assisted Decomposition of Methane and Propane and Cracking of Liquid Hexadecane
PLASMA ASSISTED DECOMPOSITION OF METHANE AND PROPANE AND CRACKING OF LIQUID HEXADECANE A thesis submitted for the degree of Doctor of Philosophy by Irma Aleknaviciute School of Engineering and Design Brunel University, Uxbridge April, 2013 Abstract Non-thermal plasmas are considered to be very promising for the initiation of chemical reactions and a vast amount of experimental work has been dedicated to plasma assisted hydrocarbon conversion processes, which are reviewed in the fourth chapter of the thesis. However, current knowledge and experimental data available in the literature on plasma assisted liquid hydrocarbon cracking and gaseous hydrocarbon decomposition is very limited. The experimental methodology is introduced in the chapter that follows the literature review. It includes the scope and objectives section reflecting the information presented in the literature review and the rationale of this work. This is followed by a thorough description of the design and construction of the experimental plasma reformer and the precise experimental procedures, the set-up of hydrocarbon characterization equipment and the development of analytical methods. The methodology of uncertainty analysis is also described. In this work we performed experiments in attempt the cracking of liquid hexadecane into smaller liquid hydrocarbons, which was not successful. The conditions tested and the problems encountered are described in detail. In this project we performed a parametric study for methane and propane decomposition under a corona discharge for CO x free hydrogen generation. For methane and propane a series of experiments were performed for a positive corona discharge at a fixed inter-electrode distance (15 mm) to study the effects of discharge power (range of 14 - 20 W and 19 – 35 W respectively) and residence time (60 - 240 s and 60 – 303 s respectively). -

Hydroisomerization of N-Hexadecane: Remarkable Selectivity of Mesoporous Silica Post- Synthetically Modified with Aluminum
Lawrence Berkeley National Laboratory Recent Work Title Hydroisomerization of n-hexadecane: Remarkable selectivity of mesoporous silica post- synthetically modified with aluminum Permalink https://escholarship.org/uc/item/6wq5s5bs Journal Catalysis Science and Technology, 7(8) ISSN 2044-4753 Authors Sabyrov, K Musselwhite, N Melaet, G et al. Publication Date 2017 DOI 10.1039/c7cy00203c Peer reviewed eScholarship.org Powered by the California Digital Library University of California Hydroisomerization of n-Hexadecane: Remarkable Selectivity of Mesoporous Silica Post-Synthetically Modified with Aluminum Kairat Sabyrov,a,c Nathan Musselwhite,a,c Gérôme Melaet,b and Gabor A. Somorjaia,b,c* aChemical Sciences Division, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, California 94720, United States. bMaterials Sciences Division, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, California 94720, United States. cDepartment of Chemistry, University of California at Berkeley, Berkeley, California 94720, United States. KEYWORDS: Platinum nanoparticles, mesoporous silica, n-hexadecane, hydroisomerization, mesoporous zeolite, bifunctional catalyst ABSTRACT: As the impact of acids on catalytically driven chemical transformations is tremendous, fundamental understanding of catalytically relevant factors is essential for the design of more efficient solid acid catalysts. In this work, we employed post-synthetic doping method to synthesize highly selective hydroisomerization catalyst and to demonstrate the effect of acid strength and density, catalyst microstructure, and platinum nanoparticle size on reaction rate and selectivity. Aluminum doped mesoporous silica catalyzed gas-phase n-hexadecane isomerization with remarkably high selectivity, producing substantially higher amount of isomers than traditional zeolite catalysts. The main reason for its high selectivity was found to be mild acidic sites generated by post-synthetic aluminum grafting. -

Viscosities and Densities of Binary Mixtures of Hexadecane With
Viscosities and Densities of Binary Mixtures of Hexadecane with Dissolved Methane or Carbon Dioxide at Temperatures From (298 to 473) K and at Pressures up to 120 MPa Malami Mohammed, Fausto Ciotta and J. P. Martin Trusler* Department of Chemical Engineering, Imperial College London, South Kensington Campus, London SW7 2AZ, U.K. * To whom correspondence should be addressed. E-mail: [email protected] ABSTRACT We report measurements of the viscosity and density of two binary mixtures comprising hexadecane with dissolved carbon dioxide or methane over the temperature range from (298.15 to 473.15) K and at pressures up to 120 MPa. The measurements were conducted at various mole fractions x of the light component as follows: x = (0, 0.0690, 0.5877 and 0.7270) for xCO2 + (1 - x)C16H34 and x = (0, 0.1013, 0.2021, 0.2976 and 0.3979) for xCH4 + (1 - x)C16H34. The viscosity and density measurements were carried out simultaneously using a bespoke vibrating-wire apparatus with a suspended sinker. With respect to the first mixture, the apparatus was operated in a relative mode and was calibrated in octane whereas, for the second mixture, the apparatus was operated in an absolute mode. To facilitate this mode of operation, the diameter of the centreless-ground tungsten wire was measured with a laser micrometer, and the mass and volume of the sinker were measured independently by hydrostatic weighing. In either mode of operation, the expanded relative uncertainties at 95 % confidence were 2 % for viscosity and 0.3 % for density. The results were correlated using simple relations that express both density and viscosity as functions of temperature and pressure. -
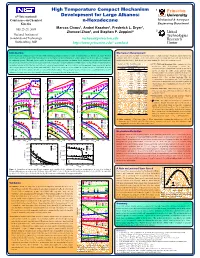
N-Hexadecane Princeton
High Temperature Compact Mechanism 1 Princeton 6th International Development for Large Alkanes: University Conference on Chemical n-Hexadecane Mechanical & Aerospace Kinetics Engineering Department 1 1 1 July 25-29, 2005 Marcos Chaos , Andrei Kazakov , Frederick L. Dryer , Zhenwei Zhao1, and Stephen P. Zeppieri2 2 United National Institute of Technologies Standards and Technology [email protected] Research Gaithersburg, MD http://www.princeton.edu/~combust Center Introduction Mechanism Development Improving internal combustion engine efficiency while minimizing pollutant emissions is a topic of continued interest. To this end, robust chemical The current mechanism was built in a hierarchal manner. A high temperature hexadecane submechanism was kinetics models and more accurate numerical tools are needed to improve our understanding of the complex physical and chemical processes taking place combined with our recent update of the H2/O2 kinetic model [3] along with C1-C4 kinetics including the in combustion systems. This study focuses on the development of a high temperature mechanism for the oxidation and pyrolisis of n-hexadecane. modifications listed in [4]. In the hexadecane submechanism, five classes of reactions are treated: Hexadecane is a primary reference fuel for cetane rating of diesel fuels and is a major component in simple mixtures used to emulate the autoignition and Table 1. H-atom abstraction rates. C-C, C-H bond homolysis: Rate constants for these combustion behavior of distillate-type fuels utilized in advanced engine designs (e.g. direct injection and homogeneous charge compression ignition (cm3-mol-s-cal) unimolecular fuel decomposition reactions were diesel engines). Models of this large hydrocarbon molecule [1, 2] are scarce and involve a large number of reactions and species which make them not Radical Log (A)/H b Ea calculated from the recombination reactions of two radical readily applicable to the simulation of multi-dimensional fluid flow problems.