Chapter 4: Diffraction Georg Roth Institute of Crystallography RWTH Aachen University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

361: Crystallography and Diffraction
361: Crystallography and Diffraction Michael Bedzyk Department of Materials Science and Engineering Northwestern University February 2, 2021 Contents 1 Catalog Description3 2 Course Outcomes3 3 361: Crystallography and Diffraction3 4 Symmetry of Crystals4 4.1 Types of Symmetry.......................... 4 4.2 Projections of Symmetry Elements and Point Groups...... 9 4.3 Translational Symmetry ....................... 17 5 Crystal Lattices 18 5.1 Indexing within a crystal lattice................... 18 5.2 Lattices................................. 22 6 Stereographic Projections 31 6.1 Projection plane............................ 33 6.2 Point of projection........................... 33 6.3 Representing Atomic Planes with Vectors............. 36 6.4 Concept of Reciprocal Lattice .................... 38 6.5 Reciprocal Lattice Vector....................... 41 7 Representative Crystal Structures 48 7.1 Crystal Structure Examples ..................... 48 7.2 The hexagonal close packed structure, unlike the examples in Figures 7.1 and 7.2, has two atoms per lattice point. These two atoms are considered to be the motif, or repeating object within the HCP lattice. ............................ 49 1 7.3 Voids in FCC.............................. 54 7.4 Atom Sizes and Coordination.................... 54 8 Introduction to Diffraction 57 8.1 X-ray .................................. 57 8.2 Interference .............................. 57 8.3 X-ray Diffraction History ...................... 59 8.4 How does X-ray diffraction work? ................. 59 8.5 Absent -

Crystal Structure of a Material Is Way in Which Atoms, Ions, Molecules Are Spatially Arranged in 3-D Space
Crystalline Structures – The Basics •Crystal structure of a material is way in which atoms, ions, molecules are spatially arranged in 3-D space. •Crystal structure = lattice (unit cell geometry) + basis (atom, ion, or molecule positions placed on lattice points within the unit cell). •A lattice is used in context when describing crystalline structures, means a 3-D array of points in space. Every lattice point must have identical surroundings. •Unit cell: smallest repetitive volume •Each crystal structure is built by stacking which contains the complete lattice unit cells and placing objects (motifs, pattern of a crystal. A unit cell is chosen basis) on the lattice points: to represent the highest level of geometric symmetry of the crystal structure. It’s the basic structural unit or building block of crystal structure. 7 crystal systems in 3-D 14 crystal lattices in 3-D a, b, and c are the lattice constants 1 a, b, g are the interaxial angles Metallic Crystal Structures (the simplest) •Recall, that a) coulombic attraction between delocalized valence electrons and positively charged cores is isotropic (non-directional), b) typically, only one element is present, so all atomic radii are the same, c) nearest neighbor distances tend to be small, and d) electron cloud shields cores from each other. •For these reasons, metallic bonding leads to close packed, dense crystal structures that maximize space filling and coordination number (number of nearest neighbors). •Most elemental metals crystallize in the FCC (face-centered cubic), BCC (body-centered cubic, or HCP (hexagonal close packed) structures: Room temperature crystal structure Crystal structure just before it melts 2 Recall: Simple Cubic (SC) Structure • Rare due to low packing density (only a-Po has this structure) • Close-packed directions are cube edges. -

Types of Lattices
Types of Lattices Types of Lattices Lattices are either: 1. Primitive (or Simple): one lattice point per unit cell. 2. Non-primitive, (or Multiple) e.g. double, triple, etc.: more than one lattice point per unit cell. Double r2 cell r1 r2 Triple r1 cell r2 r1 Primitive cell N + e 4 Ne = number of lattice points on cell edges (shared by 4 cells) •When repeated by successive translations e =edge reproduce periodic pattern. •Multiple cells are usually selected to make obvious the higher symmetry (usually rotational symmetry) that is possessed by the 1 lattice, which may not be immediately evident from primitive cell. Lattice Points- Review 2 Arrangement of Lattice Points 3 Arrangement of Lattice Points (continued) •These are known as the basis vectors, which we will come back to. •These are not translation vectors (R) since they have non- integer values. The complexity of the system depends upon the symmetry requirements (is it lost or maintained?) by applying the symmetry operations (rotation, reflection, inversion and translation). 4 The Five 2-D Bravais Lattices •From the previous definitions of the four 2-D and seven 3-D crystal systems, we know that there are four and seven primitive unit cells (with 1 lattice point/unit cell), respectively. •We can then ask: can we add additional lattice points to the primitive lattices (or nets), in such a way that we still have a lattice (net) belonging to the same crystal system (with symmetry requirements)? •First illustrate this for 2-D nets, where we know that the surroundings of each lattice point must be identical. -

The Metrical Matrix in Teaching Mineralogy G. V
THE METRICAL MATRIX IN TEACHING MINERALOGY G. V. GIBBS Department of Geological Sciences & Department of Materials Science and Engineering Virginia Polytechnic Institute and State University Blacksburg, VA 24061 [email protected] INTRODUCTION The calculation of the d-spacings, the angles between planes and zones, the bond lengths and angles and other important geometric relationships for a mineral can be a tedious task both for the student and the instructor, particularly when completed with the large as- sortment of trigonometric identities and algebraic formulae that are available (d. Crystal Geometry (1959), Donnay and Donnay, International Tables for Crystallography, Vol. II, Section 3, The Kynoch Press, 101-158). However, such calculations are straightforward and relatively easy to do when completed with the metrical matrix and the interactive software MATOP. Several applications of the matrix are presented below, each of which is worked out in detail and which is designed to teach you its use in the study of crystal geometry. SOME PRELIMINARY COMMENTS We begin our discussion of the matrix with a brief examination of the properties of the geometric three dimensional space, S, in which we live and in which minerals and rocks occur. For our purposes, it will be convenient to view S as the set of all vectors that radiate from a common origin to each point in space. In constructing a model for S, we chose three noncoplanar, coordinate axes denoted X, Y and Z, each radiating from the origin, O. Next, we place three nonzero vectors denoted a, band c along X, Y and Z, respectively, likewise radiating from O. -
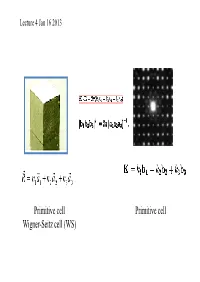
Primitive Cell Wigner-Seitz Cell (WS) Primitive Cell
Lecture 4 Jan 16 2013 Primitive cell Primitive cell Wigner-Seitz cell (WS) First Brillouin zone The Wigner-Seitz primitive cell of the reciprocal lattice is known as the first Brillouin zone. (Wigner-Seitz is real space concept while Brillouin zone is a reciprocal space idea). Powder cell Polymorphic Forms of Carbon Graphite – a soft, black, flaky solid, with a layered structure – parallel hexagonal arrays of carbon atoms – weak van der Waal’s forces between layers – planes slide easily over one another Miller indices Simple cubic Miller Indices Rules for determining Miller Indices: 1. Determine the intercepts of the face along the crystallographic axes, in terms of un it ce ll dimens ions. 2. Take the reciprocals 3. Clear fractions 4. Reduce to lowest terms Simple cubic d100=? Where does a protein crystallographer see the Miller indices? CtlCommon crystal faces are parallel to lattice planes • Eac h diffrac tion spo t can be regarded as a X-ray beam reflected from a lattice plane , and therefore has a unique Miller index. Miller indices A Miller index is a series of coprime integers that are inversely ppproportional to the intercepts of the cry stal face or crystallographic planes with the edges of the unit cell. It describes the orientation of a plane in the 3-D lattice with respect to the axes. The general form of the Miller index is (h, k, l) where h, k, and l are integers related to the unit cell along the a, b, c crystal axes. Irreducible brillouin zone II II II II Reciprocal lattice ghakblc The Bravais lattice after Fourier transform real space reciprocal lattice normaltthlls to the planes (vect ors ) poitints spacing between planes 1/distance between points ((y,p)actually, 2p/distance) l (distance, wavelength) 2p/l=k (momentum, wave number) BillBravais cell Wigner-SiSeitz ce ll Brillouin zone . -

Chapter 4, Bravais Lattice Primitive Vectors
Chapter 4, Bravais Lattice A Bravais lattice is the collection of all (and only those) points in space reachable from the origin with position vectors: n , n , n integer (+, -, or 0) r r r r 1 2 3 R = n1a1 + n2 a2 + n3a3 a1, a2, and a3 not all in same plane The three primitive vectors, a1, a2, and a3, uniquely define a Bravais lattice. However, for one Bravais lattice, there are many choices for the primitive vectors. A Bravais lattice is infinite. It is identical (in every aspect) when viewed from any of its lattice points. This is not a Bravais lattice. Honeycomb: P and Q are equivalent. R is not. A Bravais lattice can be defined as either the collection of lattice points, or the primitive translation vectors which construct the lattice. POINT Q OBJECT: Remember that a Bravais lattice has only points. Points, being dimensionless and isotropic, have full spatial symmetry (invariant under any point symmetry operation). Primitive Vectors There are many choices for the primitive vectors of a Bravais lattice. One sure way to find a set of primitive vectors (as described in Problem 4 .8) is the following: (1) a1 is the vector to a nearest neighbor lattice point. (2) a2 is the vector to a lattice points closest to, but not on, the a1 axis. (3) a3 is the vector to a lattice point nearest, but not on, the a18a2 plane. How does one prove that this is a set of primitive vectors? Hint: there should be no lattice points inside, or on the faces (lll)fhlhd(lllid)fd(parallolegrams) of, the polyhedron (parallelepiped) formed by these three vectors. -

Additive Effects of Alkali Metals on Cu-Modified Ch3nh3pbi3−Δclδ
RSC Advances PAPER View Article Online View Journal | View Issue Additive effects of alkali metals on Cu-modified CH3NH3PbI3ÀdCld photovoltaic devices† Cite this: RSC Adv.,2019,9,24231 Naoki Ueoka, Takeo Oku * and Atsushi Suzuki We investigated the addition of alkali metal elements (namely Na+,K+,Rb+, and Cs+) to Cu-modified CH3NH3PbI3ÀdCld photovoltaic devices and their effects on the photovoltaic properties and electronic structure. The open-circuit voltage was increased by CuBr2 addition to the CH3NH3PbI3ÀdCld precursor solution. The series resistance was decreased by simultaneous addition of CuBr2 and RbI, which increased the external quantum efficiencies in the range of 300–500 nm, and the short-circuit current density. The energy gap of the perovskite crystal increased through CuBr2 addition, which we also confirmed by first-principles calculations. Charge carrier generation was observed in the range of 300– Received 25th April 2019 500 nm as an increase of the external quantum efficiency, owing to the partial density of states Accepted 24th July 2019 contributed by alkali metal elements. Calculations suggested that the Gibbs energies were decreased by DOI: 10.1039/c9ra03068a incorporation of alkali metal elements into the perovskite crystals. The conversion efficiency was Creative Commons Attribution 3.0 Unported Licence. rsc.li/rsc-advances maintained for 7 weeks for devices with added CuBr2 and RbI. PbI2 +CH3NH3I + 2CH3NH3Cl / CH3NH3PbI3 Introduction + 2CH3NH3Cl (g)[ (140 C)(2) Studies of methylammonium lead halide perovskite solar x y / cells started in 2009, when a conversion efficiency of 3.9% PbI2 + CH3NH3I+ CH3NH3Cl (CH3NH3)x+yPbI2+xCly 1 / [ was reported. Some devices have since yielded conversion CH3NH3PbI3 +CH3NH3Cl (g) (140 C) (3) efficiencies of more than 20% as studies have expanded This article is licensed under a 2–5 Owing to the formation of intermediates and solvent evap- globally, with expectations for these perovskite solar cells 6–9 oration, perovskite grains are gradually formed by annealing. -

Pharmaceutico Analytical Study of Udayabhaskara Rasa
INTERNATIONAL AYURVEDIC MEDICAL JOURNAL Research Article ISSN: 2320 5091 Impact Factor: 4.018 PHARMACEUTICO ANALYTICAL STUDY OF UDAYABHASKARA RASA Gopi Krishna Maddikera1, Ranjith. B. M2, Lavanya. S. A3, Parikshitha Navada4 1Professor & HOD, Dept of Rasashastra & Bhaishajya Kalpana, S.J.G Ayurvedic Medical College & P.G Centre, Koppal, Karnataka, India 2Ayurveda Vaidya; 3Ayurveda Vaidya; 43rd year P.G Scholar, T.G.A.M.C, Bellary, Karnataka, India Email: [email protected] Published online: January, 2019 © International Ayurvedic Medical Journal, India 2019 ABSTRACT Udayabhaskara rasa is an eccentric formulation which is favourable in the management of Amavata. Regardless of its betterment in the management of Amavata, no research work has been carried out till date. The main aim of this study was preparation of Udayabhaskara Rasa as disclosed in the classics & Physico-chemical analysis of Udayabhaskara Rasa. Udayabhaskara Rasa was processed using Kajjali, Vyosha, dwikshara, pancha lavana, jayapala and beejapoora swarasa. The above ingredients were mixed to get a homogenous mixture of Udayab- haskara Rasa which was given 1 Bhavana with beejapoora swarasa and later it is dried and stored in air-tight container. The Physico chemical analysis of Udayabhaskara Rasa before (UB-BB) and after bhavana (UB-AB) was done. Keywords: Udayabhaskara Rasa, XRD, FTIR, SEM-EDAX. INTRODUCTION Rasashastra is a branch of Ayurveda which deals with Udayabhaskara Rasa. In the present study the formu- metallo-mineral preparations aimed at achieving Lo- lation is taken from the text brihat nighantu ratna- havada & Dehavada. These preparations became ac- kara. The analytical study reveals the chemical com- ceptable due to its assimilatory property in the minute position of the formulations as well as their concentra- doses. -

Symmetry and Groups, and Crystal Structures
CHAPTER 3: SYMMETRY AND GROUPS, AND CRYSTAL STRUCTURES Sarah Lambart RECAP CHAP. 2 2 different types of close packing: hcp: tetrahedral interstice (ABABA) ccp: octahedral interstice (ABCABC) Definitions: The coordination number or CN is the number of closest neighbors of opposite charge around an ion. It can range from 2 to 12 in ionic structures. These structures are called coordination polyhedron. RECAP CHAP. 2 Rx/Rz C.N. Type Hexagonal or An ideal close-packing of sphere 1.0 12 Cubic for a given CN, can only be Closest Packing achieved for a specific ratio of 1.0 - 0.732 8 Cubic ionic radii between the anions and 0.732 - 0.414 6 Octahedral Tetrahedral (ex.: the cations. 0.414 - 0.225 4 4- SiO4 ) 0.225 - 0.155 3 Triangular <0.155 2 Linear RECAP CHAP. 2 Pauling’s rule: #1: the coodination polyhedron is defined by the ratio Rcation/Ranion #2: The Electrostatic Valency (e.v.) Principle: ev = Z/CN #3: Shared edges and faces of coordination polyhedra decreases the stability of the crystal. #4: In crystal with different cations, those of high valency and small CN tend not to share polyhedral elements #5: The principle of parsimony: The number of different sites in a crystal tends to be small. CONTENT CHAP. 3 (2-3 LECTURES) Definitions: unit cell and lattice 7 Crystal systems 14 Bravais lattices Element of symmetry CRYSTAL LATTICE IN TWO DIMENSIONS A crystal consists of atoms, molecules, or ions in a pattern that repeats in three dimensions. The geometry of the repeating pattern of a crystal can be described in terms of a crystal lattice, constructed by connecting equivalent points throughout the crystal. -

PHZ6426 Crystal Structure
SYMMETRY Finite geometric objects (“molecules) are symmetric with respect to a) rotations b) reflections (symmetry planes, symmetry points) c) combinatons of translations and rotations (screw axes) 2π If an object is symmetric with respect to rotation by angle φ = , it is said to have has an “n-fold rotational axis” n n=1 is a trivial case: every object is symmetric about a full revolution. Non-trivial axes have n>1. 1 Symmetries of an equilateral triangle 2 2 2 180-rotations (flips) 3’ 1’ about 22’ 3 1 3 1 2’ about 33’ about 11’ 1 3 1 2 2 3 3 2 2π 1 120 degree= rotations 3 3 2 1 3 3 3 4 2 2 1 1 An equilateral triangle is not symmetric with respect to inversion about its center. But a square is. 5 Gliding (screw) axis Rotate by 180 degrees about the axis Slide by a half-period 6 2-fold rotations can be viewed as reflections in a plane which contains a 2-fold rotation axis 7 Crystal Structure Crystal structure can be obtained by attaching atoms or groups of atoms --basis-- to lattice sites. Crystal Structure = Crystal Lattice + Basis Crystal Structure 88 Partially from Prof. C. W. Myles (Texas Tech) course presentation Building blocks of lattices are geometric figures with certain symmetries: rotational, reflectional, etc. Lattice must obey all these symmetries but, in addition, it also must obey TRANSLATIONAL symmetry Which geometric figures can be used to built a lattice? 9 10 Crystallographic Restriction Theorem: Crystal lattices have only 2-,3, 4-, and 6-fold rotational symmetries 11 B’ ma A’ n-fold axis φ-π/2 φ=2π/n a A B 1. -

Lattice Vibrations - Phonons
Lattice vibrations - phonons So far, we have assumed that the ions are fixed at their equilibrium positions, and we focussed on understanding the motion of the electrons in the static periodic potential created by the ions. But, of course, the ions are quantum objects that cannot be at rest in well-defined positions { this would violate Heisenberg's uncertainty principle. So they must be moving! In the following, we will assume that we are at temperatures very well below the melting temperature of the crystal, so that the ions perform small oscillations about their equilibrium positions. Of course, with increasing T the amplitude of these vibrations increases and eventually becomes large enough that the ions start moving past one another and the lattice order is progressively lost (the crystal starts to melt); our approximations will not be valid once we reach such high temperatures. Dealing with small oscillations is very nice (as you may remember from studying Lagrangian/Hamiltonian dynamics) because in the harmonic approximation, i.e. where we expand potentials up to second- order in these displacements, such classical problems can be solved exactly by mapping them to a sum of independent harmonic oscillators. Going to the quantum solution is then trivial, because it will simply be the sum of independent quantum harmonic oscillators. In this chapter we dis- cuss this solution and its implications, and for the time being we will completely forget about the electrons. We will return back to them in the next section. I assume you have already studied phonons in your undergraduate solid state physics course. -

An Atomistic Study of Phase Transition in Cubic Diamond Si Single Crystal T Subjected to Static Compression ⁎ Dipak Prasad, Nilanjan Mitra
Computational Materials Science 156 (2019) 232–240 Contents lists available at ScienceDirect Computational Materials Science journal homepage: www.elsevier.com/locate/commatsci An atomistic study of phase transition in cubic diamond Si single crystal T subjected to static compression ⁎ Dipak Prasad, Nilanjan Mitra Indian Institute of Technology Kharagpur, Kharagpur 721302, India ARTICLE INFO ABSTRACT Keywords: It is been widely experimentally reported that Si under static compression (typically in a Diamond Anvil Setup- Molecular dynamics DAC) undergoes different phase transitions. Even though numerous interatomic potentials are used fornu- Phase transition merical studies of Si under different loading conditions, the efficacy of different available interatomic potentials Hydrostatic and Uniaxial compressive loading in determining the phase transition behavior in a simulation environment similar to that of DAC has not been Cubic diamond single crystal Silicon probed in literature which this manuscript addresses. Hydrostatic compression of Silicon using seven different interatomic potentials demonstrates that Tersoff(T0) performed better as compared to other potentials with regards to demonstration of phase transition. Using this Tersoff(T0) interatomic potential, molecular dynamics simulation of cubic diamond single crystal silicon has been carried out along different directions under uniaxial stress condition to determine anisotropy of the samples, if any. -tin phase could be observed for the [001] direction loading whereas Imma along with -tin phase could be observed for [011] and [111] direction loading. Amorphization is also observed for [011] direction. The results obtained in the study are based on rigorous X-ray diffraction analysis. No strain rate effects could be observed for the uniaxial loading conditions. 1. Introduction potential(SW) [19,20] for their simulations.