Identification of Epigenetic Mechanisms Involved in the Anti-Asthmatic Effects of Descurainia Sophia Seed Extract Based on a Multi-Omics Approach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cell Death Via Lipid Peroxidation and Protein Aggregation Diseases
biology Review Cell Death via Lipid Peroxidation and Protein Aggregation Diseases Katsuya Iuchi * , Tomoka Takai and Hisashi Hisatomi Department of Materials and Life Science, Faculty of Science and Technology, Seikei University, 3-3-1 Kichijojikitamachi, Musashino-shi, Tokyo 180-8633, Japan; [email protected] (T.T.); [email protected] (H.H.) * Correspondence: [email protected] or [email protected]; Tel.: +81-422-37-3523 Simple Summary: It is essential for cellular homeostasis that biomolecules, such as DNA, proteins, and lipids, function properly. Disturbance of redox homeostasis produces aberrant biomolecules, including oxidized lipids and misfolded proteins, which increase in cells. Aberrant biomolecules are removed by excellent cellular clearance systems. However, when excess aberrant biomolecules remain in the cell, they disrupt organelle and cellular functions, leading to cell death. These aberrant molecules aggregate and cause apoptotic and non-apoptotic cell death, leading to various protein aggregation diseases. Thus, we investigated the cell-death cross-linking between lipid peroxidation and protein aggregation. Abstract: Lipid peroxidation of cellular membranes is a complicated cellular event, and it is both the cause and result of various diseases, such as ischemia-reperfusion injury, neurodegenerative diseases, and atherosclerosis. Lipid peroxidation causes non-apoptotic cell death, which is associated with cell fate determination: survival or cell death. During the radical chain reaction of lipid peroxidation, Citation: Iuchi, K.; Takai, T.; various oxidized lipid products accumulate in cells, followed by organelle dysfunction and the Hisatomi, H. Cell Death via Lipid induction of non-apoptotic cell death. Highly reactive oxidized products from unsaturated fatty acids Peroxidation and Protein are detected under pathological conditions. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Regulation of Mitochondrial and Nonmitochondrial Protein Turnover by the PINK1-Parkin Pathway
Regulation of mitochondrial and nonmitochondrial protein turnover by the PINK1-Parkin pathway Evelyn S. Vincow A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2013 Reading Committee: Leo J. Pallanck, Chair Sandra M. Bajjalieh Michael J. MacCoss Program Authorized to Offer Degree: Neurobiology and Behavior © Copyright 2013 Evelyn S. Vincow University of Washington Abstract Regulation of mitochondrial and nonmitochondrial protein turnover by the PINK1-Parkin pathway Evelyn Sandra Vincow Chair of the Supervisory Committee: Associate Professor Leo J. Pallanck Genome Sciences The accumulation of damaged mitochondria has been proposed as a key factor in aging and in the pathogenesis of many common age-related diseases, including Parkinson disease (PD). Recently, in vitro studies of the PD-related proteins Parkin and PINK1 have found that these factors act in a common pathway to promote the selective autophagic degradation of damaged mitochondria (mitophagy). However, whether PINK1 and Parkin promote mitophagy in vivo is unknown. To address this question, I used a proteomic approach in Drosophila to study the effects of null mutations in parkin or PINK1 on mitochondrial protein turnover. The parkin null mutants showed a significant overall slowing of mitochondrial protein turnover, similar to but less severe than the slowing seen in autophagy-deficient Atg7 mutants, consistent with the model that Parkin acts upstream of Atg7 to promote mitophagy. By contrast, the turnover of many mitochondrial respiratory chain (RC) subunits showed greater impairment in parkin than in Atg7 mutants, and RC turnover was also selectively impaired in PINK1 mutants. These findings demonstrate that the PINK1-Parkin pathway promotes mitophagy in vivo and, unexpectedly, also promotes selective turnover of mitochondrial RC components. -

An ACSL4 Hemizygous Intragenic Deletion in a Patient with Childhood Stroke
Pediatric Neurology 100 (2019) 100e101 Contents lists available at ScienceDirect Pediatric Neurology journal homepage: www.elsevier.com/locate/pnu Clinical Letter An ACSL4 Hemizygous Intragenic Deletion in a Patient With Childhood Stroke Caitlin A. Chang, MD a, Julie Lauzon, MD, MHSc a, b, Adam Kirton, MD, MSc b, c, * Bob Argiropoulos, PhD a, b, d, a Department of Medical Genetics, Alberta Children's Hospital, Calgary, Alberta, Canada b Alberta Children's Hospital Research Institute for Child and Maternal Health, Alberta Children's Hospital, Calgary, Alberta, Canada c Department of Pediatrics and Clinical Neurosciences, Pediatric Neurology, Alberta Children's Hospital, Calgary, Alberta, Canada d Genetic Laboratory Services, Cytogenetics Laboratory, Alberta Children's Hospital, Calgary, Alberta, Canada article info Article history: Received 4 March 2019 Accepted 22 June 2019 Available online 28 June 2019 Keywords: Childhood stroke ACSL4 X-linked intellectual disability Ischemia Developmental delay We describe a male with a maternally inherited 9.75-kb intra- was present. Additional evaluation for stroke risk factors, including genic deletion of ACSL4. Fewer than 25 individuals have been re- lumbar puncture, echocardiogram, and examination of blood for fl ported with ACSL4-related X-linked intellectual disability (XLID). thrombophilia and in ammatory markers were negative. This individual highlights a unique presentation of ACSL4-related At three years, severe communication delays were evident. fi XLID and reveals additional pathways for investigation. Behavioral dif culties included frequent outbursts, irritability, and tantrums. There were no limitations in gross or fine motor func- Patient description tioning. At 4.5 years, he developed seizures characterized by eye deviation and automatisms. -

Supplementary Material
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Page 1 / 45 SUPPLEMENTARY MATERIAL Appendix A1: Neuropsychological protocol. Appendix A2: Description of the four cases at the transitional stage. Table A1: Clinical status and center proportion in each batch. Table A2: Complete output from EdgeR. Table A3: List of the putative target genes. Table A4: Complete output from DIANA-miRPath v.3. Table A5: Comparison of studies investigating miRNAs from brain samples. Figure A1: Stratified nested cross-validation. Figure A2: Expression heatmap of miRNA signature. Figure A3: Bootstrapped ROC AUC scores. Figure A4: ROC AUC scores with 100 different fold splits. Figure A5: Presymptomatic subjects probability scores. Figure A6: Heatmap of the level of enrichment in KEGG pathways. Kmetzsch V, et al. J Neurol Neurosurg Psychiatry 2021; 92:485–493. doi: 10.1136/jnnp-2020-324647 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Appendix A1. Neuropsychological protocol The PREV-DEMALS cognitive evaluation included standardized neuropsychological tests to investigate all cognitive domains, and in particular frontal lobe functions. The scores were provided previously (Bertrand et al., 2018). Briefly, global cognitive efficiency was evaluated by means of Mini-Mental State Examination (MMSE) and Mattis Dementia Rating Scale (MDRS). Frontal executive functions were assessed with Frontal Assessment Battery (FAB), forward and backward digit spans, Trail Making Test part A and B (TMT-A and TMT-B), Wisconsin Card Sorting Test (WCST), and Symbol-Digit Modalities test. -

Identification of ACSL4 As a Biomarker and Contributor of Ferroptosis In
Identication of ACSL4 as a biomarker and contributor of ferroptosis in clear cell renal cell carcinoma na guo ( [email protected] ) Primary research Keywords: Acyl-CoA synthetase long-chain family member 4 (ACSL4), biomarker, ferroptosis, clear cell renal cell carcinoma (ccRCC) Posted Date: May 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-24079/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/17 Abstract Background ACSL4 has been reported to be related to tumor genesis and involved in the processes of ferroptosis. However, the expression levels and prognostic value of ACSL4 in clear cell renal cell carcinoma (ccRCC) remain unclear. Methods The Oncomine and TCGA databases were used to predict the expression of ACSL4 mRNA in ccRCC and its association with ccRCC prognosis. The expression levels of ACSL4 were determined in human RCC tissues by real-time PCR. Kaplan-Meier curves were used to analyze the diagnostic and prognostic signicance of ACSL4 in ccRCC. A ferroptosis inducer (erastin) was used to investigate the effects of ACSL4 on ferroptosis in ccRCC cell lines. Results The expression level of ACSL4 was signicantly down-regulated in ccRCC tissues (P < 0.001), which was consistent with the analysis of the Oncomine and TCGA database. Then, immunohistochemical results demonstrated that the ACSL4 was weak or not detected in ccRCC tissues than that in normal tissues. ACSL4 differential expression level was signicantly related to gender, ccRCC subtypes, nodal invasion, tumor grade and cancer stages (all P < 0.001). Survival analysis revealed that overall survival was favorable in ccRCC patients with ACSL4 high expression (P = 0.014). -

Molecular Signature Induced by RNASET2, a Tumor Antagonizing Gene, in Ovarian Cancer Cells
www.impactjournals.com/oncotarget/ Oncotarget, June, Vol.2, No 6 Molecular signature induced by RNASET2, a tumor antagonizing gene, in ovarian cancer cells Francesco Acquati1, Laura Monti1, Marta Lualdi1, Marco Fabbri2, Maria Grazia Sacco2, Laura Gribaldo2, and Roberto Taramelli1 1 Dipartimento di Biotecnologie e Scienze Molecolari, Università degli Studi dell’Insubria, via JH Dunant 3, 21100 Varese, Italy 2 European Commission - Joint Research Centre Institute for Health and Consumer Protection Molecular Biology and Genomics unit TP 464, Via E. Fermi, 2749 21027 Ispra (VA) - Italy Correspondence to: Roberto Taramelli, email: [email protected] Correspondence to: Francesco Acquati, email: [email protected] Keywords: RNases, cancer microenvironment, transcriptional profile Received: May 10, 2011, Accepted: June 2, 2011, Published: June 4, 2011 Copyright: © Acquati et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT: Using the Hey3Met2 human ovarian cancer cell line, we previously found the RNASET2 gene to possess a remarkable in vivo tumor suppressor activity, although no in vitro features such as inhibition of cell proliferation, clonogenic potential, impaired growth in soft agar and increase in apoptotic rate could be detected. This is reminiscent of the behavior of genes belonging to the class of tumor antagonizing genes (TAG) which act mainly within the context of the microenvironment. Here we present transcriptional profiles analysis which indicates that investigations of the mechanisms of TAG biological functions require a comparison between the in vitro and in vivo expression patterns. -

Global Ubiquitylation Analysis of Mitochondria in Primary Neurons
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.01.438131; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Global ubiquitylation analysis of mitochondria in primary neurons identifies physiological Parkin targets following activation of PINK1 Odetta Antico1#, Alban Ordureau2#, Michael Stevens1, Francois Singh1, Marek φ Gierlinski3, Erica Barini1 , Mollie L. Rickwood1, Alan Prescott4, Rachel Toth1, Ian G. Ganley1, J. Wade Harper2*, and Miratul M. K. Muqit1* 1MRC Protein Phosphorylation and Ubiquitylation Unit, School of Life Sciences, University of Dundee, Dundee, United Kingdom, DD1 5EH, U.K. 2Department of Cell Biology, Harvard Medical School, Boston, MA 02115, USA 3Data Analysis Group, Division of Computational Biology, School of Life Sciences, University of Dundee, Dundee, United Kingdom, DD1 5EH, U.K. 4Dundee Imaging Facility, School of Life Sciences, University of Dundee, Dundee, United Kingdom, DD1 5EH, U.K #Denotes Equal Contribution φCurrent address: AbbVie Deutschland GmbH & Co, Knollstr, 67061, Ludwigshafen, Germany *Correspondence: [email protected] or [email protected] Keywords Neurons, PINK1, Parkin, ubiquitin, Parkinson’s disease, Mitochondria, Running Title: Ubiquitin analysis of mitochondria in neurons 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.04.01.438131; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
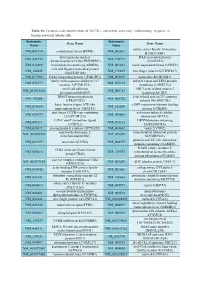
Systematic Name Gene Name Systematic Name Gene Name NM 001710 Complement Factor B(CFB) NM 052831 Solute Carrier Family 18 Member
Table S1: Genome-wide identification of SGLT2i`s interaction with early inflammatory response in human proximal tubular cells. Systematic Systematic Gene Name Gene Name Name Name solute carrier family 18 member NM_001710 complement factor B(CFB) NM_052831 B1(SLC18B1) heterogeneous nuclear DAZ associated protein NM_031372 NM_170711 ribonucleoprotein D like(HNRNPDL) 1(DAZAP1) NM_014299 bromodomain containing 4(BRD4) NM_001261 cyclin dependent kinase 9(CDK9) cilia and flagella associated protein NM_182628 NM_178835 zinc finger protein 827(ZNF827) 100(CFAP100) NM_017906 PAK1 interacting protein 1(PAK1IP1) NM_024015 homeobox B4(HOXB4) family with sequence similarity 167 ankyrin repeat and LEM domain NM_053279 NM_015114 member A(FAM167A) containing 2(ANKLE2) small cell adhesion ARP3 actin related protein 3 NM_001031628 NM_005721 glycoprotein(SMAGP) homolog(ACTR3) TRAF3 interacting protein actin related protein 2/3 complex NM_147686 NM_005720 2(TRAF3IP2) subunit 1B(ARPC1B) basic leucine zipper ATF-like cAMP responsive element binding NM_018664 NM_182898 transcription factor 3(BATF3) protein 5(CREB5) zinc finger CCCH-type containing activation induced cytidine NM_025079 NM_020661 12A(ZC3H12A) deaminase(AICDA) C-X-C motif chemokine ligand DENN domain containing NM_001511 NM_015213 1(CXCL1) 5A(DENND5A) NM_025072 prostaglandin E synthase 2(PTGES2) NM_004665 vanin 2(VNN2) superoxide dismutase 2, mitochondrial ribosomal protein NM_001024465 NM_016070 mitochondrial(SOD2) S23(MRPS23) jumonji and AT-rich interaction NM_033199 urocortin 2(UCN2) NM_004973 -

Therapeutic Vaccines for Amyotrophic Lateral Sclerosis Directed Against
Vaccine 37 (2019) 4920–4927 Contents lists available at ScienceDirect Vaccine journal homepage: www.elsevier.com/locate/vaccine Therapeutic vaccines for amyotrophic lateral sclerosis directed against disease specific epitopes of superoxide dismutase 1 ⇑ Beibei Zhao a, Kristen Marciniuk b,c, Ebrima Gibbs a, Masoud Yousefi a, Scott Napper b,c, Neil R. Cashman a, a Djavad Mowafaghian Centre for Brain Health, University of British Columbia, Vancouver, BC V6T 2B5, Canada b Department of Biochemistry, Microbiology, and Immunology, University of Saskatchewan, Saskatoon, Saskatchewan S7N 5E5, Canada c Vaccine and Infectious Disease Organization International Vaccine Research Center, University of Saskatchewan, Saskatchewan, SK S7N 5E3, Canada article info abstract Article history: Emerging evidence suggests seeding and prion-like propagation of mutant Superoxide Dismutase 1 Received 6 February 2019 (SOD1) misfolding to be a potential mechanism for ALS pathogenesis and progression. Immuno- Received in revised form 8 June 2019 targeting of misfolded SOD1 has shown positive clinical outcomes in mutant SOD1 transgenic mice. Accepted 10 July 2019 However, a major challenge in developing active immunotherapies for proteinopathies such as ALS is Available online 16 July 2019 the design of immunogens enabling exclusive recognition of pathogenic species of a self-protein. Ideally, one would achieve a robust antibody response against the disease-misfolded protein while spar- ing the natively folded conformer to avoid inducing deleterious autoimmune complications, or inhibiting its normal function. Using a motor neuron disease mouse model expressing human SOD1-G37R, we herein report the immunogenicity and therapeutic efficacy of two ALS vaccines, tgG-DSE2lim and tgG- DSE5b, based on the notion that native SOD1 would undergo early unfolding in disease to present ‘‘dis- ease specific epitopes” (DSE). -

Links Between Iron and Lipids: Implications in Some Major Human Diseases
pharmaceuticals Review Links Between Iron and Lipids: Implications in Some Major Human Diseases Stephanie Rockfield, Ravneet Chhabra, Michelle Robertson, Nabila Rehman, Richa Bisht and Meera Nanjundan * Department of Cell Biology, Microbiology and Molecular Biology, University of South Florida, Tampa, FL 336200, USA; srockfi[email protected] (S.R.); [email protected] (R.C.); [email protected] (M.R.); [email protected] (N.R.); [email protected] (R.B.) * Correspondence: [email protected]; Tel.: +1-813-974-8133 Received: 31 August 2018; Accepted: 19 October 2018; Published: 22 October 2018 Abstract: Maintenance of iron homeostasis is critical to cellular health as both its excess and insufficiency are detrimental. Likewise, lipids, which are essential components of cellular membranes and signaling mediators, must also be tightly regulated to hinder disease progression. Recent research, using a myriad of model organisms, as well as data from clinical studies, has revealed links between these two metabolic pathways, but the mechanisms behind these interactions and the role these have in the progression of human diseases remains unclear. In this review, we summarize literature describing cross-talk between iron and lipid pathways, including alterations in cholesterol, sphingolipid, and lipid droplet metabolism in response to changes in iron levels. We discuss human diseases correlating with both iron and lipid alterations, including neurodegenerative disorders, and the available evidence regarding the potential mechanisms underlying how iron may promote disease pathogenesis. Finally, we review research regarding iron reduction techniques and their therapeutic potential in treating patients with these debilitating conditions. We propose that iron-mediated alterations in lipid metabolic pathways are involved in the progression of these diseases, but further research is direly needed to elucidate the mechanisms involved. -

A Genome Wide Association Study of Fast Beta EEG in Families of European Ancestry
CORE Metadata, citation and similar papers at core.ac.uk Provided by IUPUIScholarWorks HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Int J Psychophysiol Manuscript Author . Author Manuscript Author manuscript; available in PMC 2018 May 01. Published in final edited form as: Int J Psychophysiol. 2017 May ; 115: 74–85. doi:10.1016/j.ijpsycho.2016.12.008. A Genome Wide Association Study of Fast Beta EEG in Families of European Ancestry Jacquelyn L. Meyers1,*, Jian Zhang1, Niklas Manz1,2, Madhavi Rangaswamy3, Chella Kamarajan1, Leah Wetherill4, David B. Chorlian1, Sun J. Kang5, Lance Bauer6, Victor Hesselbrock6, John Kramer7, Samuel Kuperman7, John I. Nurnberger Jr4, Jay Tischfield8, Jen Chyong Wang9, Howard J. Edenberg4,10, Alison Goate9, Tatiana Foroud4, and Bernice Porjesz1 1Henri Begleiter Neurodynamics Laboratory, Department of Psychiatry and Behavioral Sciences, SUNY Downstate Medical Center, Brooklyn, NY, USA 2Department of Physics, College of Wooster, Wooster, OH, USA 3Department of Psychology, Christ University, Bangalore, India 4Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, USA 5Albany Stratton VA Medical Center, Albany, NY, USA 6University of Connecticut School of Medicine, Farmington, CT, USA 7Department of Psychiatry, University of Iowa, Iowa City, IA, USA 8Rutgers University, Piscataway, NJ, USA 9Icahn School of Medicine at Mt. Sinai, New York, NY, USA 10Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, IN, USA Abstract BACKGROUND—Differences in fast beta (20–28 Hz) electroencephalogram (EEG) oscillatory activity distinguish some individuals with psychiatric and substance use disorders, suggesting that it may be a useful endophenotype for studying the genetics of disorders characterized by neural hyper-excitability.