Characterization of Bacterial Microbiota Composition Along the Gastrointestinal Tract in Rabbits
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Shafan – Hyrax Or Rabbit? Jonathan S
Shafan – Hyrax or Rabbit? Jonathan S. Ostroff, 26 Ellul 5773 Revised version of article appearing in Dialogue, Fall 5774, No. 4 Could the shafan be the rabbit? R. Slifkin’s answer is no. He concedes that many Rishonim understood the shafan to be the rab- bit, but summarily dismisses their position. He claims that, as Europeans, the Rishonim were una- ware of the fauna of the Middle East. On his blog R. Slifkin writes: The original study was by Tchernov [2000], who notes that the hare is “the only endemic species of lagomorph known from the Middle East since the Middle Pleistocene”. 1 Lagomorphs include hares, rabbits and pikas. The study by Tchernov et. al. claims that hare remains have been found in the Middle East, but not the remains of rabbits. In addition, according to R. Slifkin, early authorities such as Rav Saadia Gaon (who lived in the Middle East) and Ibn Janach (about 100 years later) identified the shafan as the hyrax. Traditional sources for identifying the shafan as the hyrax include Rav Saadia Gaon (882-924CE), Ibn Janach and Tevuos Ha-Aretz. [N. Slifkin, The Camel, the Hare and the Hyrax, p88, 2011, 2nd edition] Accordingly, R. Slifkin claims that the shafan is definitively the hyrax. Even though the hyrax does not regurgitate its food, the Torah calls it ma'aleh geira because its chewing motion superficially resembles that of ruminants, even though the chewing action is not needed for nutrition. R. Slifkin’s interpretation is somewhat puzzling.2 If the hyrax is not actually ma’alah geira, why is it so described? Would it not be more reasonable for the Torah to disabuse people of this fiction and explain that the reason the hyrax is not kosher is because it is neither split-hooved nor ma’alei geirah? This fictional criterion also poses a problem as it would apply to other animals not mentioned in the Torah’s exhaustive list (e.g. -

Hand Raising Domestic Baby Rabbits a Practical Manual
Hand Raising Domestic Baby Rabbits A practical manual Chris Mathyssek, PhD with Ciarra Rawleigh, BS Table of Contents Preface .......................................................................................................................................................... 3 About this manual ......................................................................................................................................... 4 First things first ............................................................................................................................................. 5 A real bunny mom as foster? .................................................................................................................... 6 The Right Environment: Where & how to house the babies? ...................................................................... 7 Living space - box, carrier or cage ............................................................................................................. 7 Cleaning the carrier/cage .......................................................................................................................... 8 Providing the right environment: Temperature, light, noise and smells .................................................. 8 Example: my set-up ................................................................................................................................. 11 Adding a Playground & Enrichment ....................................................................................................... -

Moringa Diet for Rabbits
EFFECT OF MORINGA LEAF MEAL (MOLM) ON NUTRIENT DIGESTIBILITY, GROWTH, CARCASS AND BLOOD INDICES OF WEANER RABBITS BY FREDERICK NUHU FEBRUARY, 2010 KWAME NKRUMAH UNIVERSITY OF SCIENCE AND TECHNOLOGY, KUMASI FACULTY OF AGRICULTURE AND NATURAL RESOURCES DEPARTMENT OF ANIMAL SCIENCE EFFECT OF MORINGA LEAF MEAL (MOLM) ON NUTRIENT DIGESTIBILITY, GROWTH, CARCASS AND BLOOD INDICES OF WEANER RABBITS A THESIS SUBMITTED TO THE SCHOOL OF GRADUATE STUDIES, KWAME NKRUMAH UNIVERSITY OF SCIENCE AND TECHNOLOGY, KUMASI, IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE AWARD OF MASTER OF SCIENCE DEGREE IN ANIMAL NUTRITION BY FREDERICK NUHU B.SC. (HONS) AGRIC. (CAPE COAST) FEBRUARY, 2010 CERTIFICATION I, Nuhu Frederick, hereby certify that the work herein submitted as a thesis for the Master of Science (Animal Nutrition) degree has neither in whole nor in part been presented nor is being concurrently submitted for any other degree elsewhere. However, works of other researchers and authors which served as sources of information were duly acknowledged by references of the authors. Name of student: Frederick Nuhu Signature: Date: Name of supervisor: Mr. P.K. Karikari Name of head of department: Prof. E.L.K. Osafo Signature: Signature: Date: Date: i TABLE OF CONTENTS Title Page Certification i List of Tables vi Acknowledgement xi Abstract xii Chapter one 1 1.0 Introduction 1 Chapter two 4 2.0 Literature Review 4 2.1 Origin and distribution of moringa 4 2.2 Uses of moringa 4 2.3 Nutritive value of moringa plant 5 2.4 Phytochemicals of moringa and their uses -

Impact of Helminth Infections and Nutritional Constraints on the Small Intestine Microbiota
RESEARCH ARTICLE Impact of Helminth Infections and Nutritional Constraints on the Small Intestine Microbiota Isabella M. Cattadori1,2*, Aswathy Sebastian3☯, Han Hao4☯, Robab Katani5☯, Istvan Albert3, Kirsten E. Eilertson4, Vivek Kapur1,5, Ashutosh Pathak1,2¤, Susan Mitchell1,2 1 Center for Infectious Disease Dynamics, The Pennsylvania State University, University Park, 16082 PA, United States of America, 2 Department of Biology, The Pennsylvania State University, University Park, 16082 PA, United States of America, 3 Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, 16082 PA, United States of America, 4 Department of a11111 Statistics, The Pennsylvania State University, University Park, 16082 PA, United States of America, 5 Department of Veterinary and Biomedical Sciences, The Pennsylvania State University, University Park, 16082 PA, United States of America ☯ These authors contributed equally to this work. ¤ Current address: Department of Infectious Diseases, College of Veterinary Medicine, The University of Georgia, Athens, GA 30602, United States of America * [email protected] OPEN ACCESS Citation: Cattadori IM, Sebastian A, Hao H, Katani R, Albert I, Eilertson KE, et al. (2016) Impact of Helminth Abstract Infections and Nutritional Constraints on the Small Intestine Microbiota. PLoS ONE 11(7): e0159770. Helminth infections and nutrition can independently alter the composition and abundance of doi:10.1371/journal.pone.0159770 the gastrointestinal microbiota, however, their combined effect is poorly understood. Here, Editor: Emmanuel Serrano Ferron, Universidade de we used the T. retortaeformis-rabbit system to examine how the helminth infection and host Aveiro, PORTUGAL restriction from coprophagy/ready-to-absorb nutrients affected the duodenal microbiota, Received: February 22, 2016 and how these changes related to the acquired immune response at the site of infection. -

Small Mammals
Self-Assessment Colour Review of Small Mammals Susan A. Brown DVM Midwest Bird & Exotic Animal Hospital Westchester, Illinois Karen L. Rosenthal MS, DVM, Dipl ABVP Avian The Animal Medical Center New York, New York Manson Publishing/The Veterinary Press Common name Scientific name Original range 13-lined ground squirrel Citellus tridecemlineatus North America African hedgehog Atelerix albiventris Africa Chinchilla Chinchilla laniger South America Common marmoset Callithrix jacchus South America Cotton-tail rabbit Sylvilagus nuttalii North America Cotton-top tamarin Saguinus oedipus South America Domestic rabbit Oryctolagus cuniculus Worldwide Eastern gray squirrel Sciurus carolinensis North America Eastern red squirrel Tamiasciurus hudsonicus North America European hedgehog Erinaceus europaeus Europe Ferret Mustela putorius furo Europe/North Africa Golden hamster Mesocricetus auratus East Europe/Middle East Guinea pig Cavia porcellus South America Lion tamarin Leontopithecus rosalia Brazil Long tailed macaque Maccaca fascicularis South-East Asia Mongolian gerbil Meriones unguiculatus Mongolia Mouse Mus musculus Worldwide Owl monkey Aotus trivirgatus Central/South America Patas monkey Erythrocebus patas Africa Potbellied pig Sus scrota Asia Prairie dog Cynomys ludovicianus North America Raccoon Procyon lotor North America Rat Rattus norvegicus Worldwide Red fox Vulpes vulpes North America Rhesus monkey Macaca mulatta India Spider monkey Ateles geoffroyi South America Squirrel monkey Saimiri sciureus South America Stripped skunk Mephitis -

Abstracts American Dairy Science Association American Society of Animal Science
ABSTRACTS AMERICAN DAIRY SCIENCE ASSOCIATION AMERICAN SOCIETY OF ANIMAL SCIENCE July 24–28, 2000 Baltimore, Maryland * Author Presenting Paper OPENING SESSION ABSTRACTS 2000 Innovation in transferring research into practice. 2001 Governance of innovation in animal production: B.D. Moser*, The Ohio State University. New roles for science, business and the public sector. G. van Dijk*1 and P.W.J. van Boekel2, 1Wageningen University, The Nether- 2 Stakeholder and governmental support for research programs is vital. lands, Nyenrode University, The Netherlands. The best way to garner support: find out what the public wants, deliver that to them, and then let them know that it’s delivered. This is espe- Innovation results from a variety of sources. Although lacking well- cially important given that most of our constituents are no longer food defined and testable theories it is widely held that systematic research producers. almost linearly translates into innovation and enhanced efficiency. The type of innovation effectuated by a system of experimental stations, re- County-based councils and advisory groups are one way to achieve this. search institutes and universities, all with specific core competences, Ongoing and regular discussions with stakeholders through these groups has shown itself as predominantly incremental: improving performance can alert colleges to current needs so that they can tailor work and es- on individual supply chain levels. In this paper, however, it is shown tablish Extension and research priorities for the coming year. Another that market orientation overthrows existing models of innovation: cer- successful model is a high-level advisory council composed of opinion tain market conditions require explorative market learning (e.g. -

Rabbit 1 Rabbit
Rabbit 1 Rabbit Rabbit Scientific classification Kingdom: Animalia Phylum: Chordata Subphylum: Vertebrata Class: Mammalia Order: Lagomorpha Family: Leporidae in part Genera Pentalagus Brachylagus Bunolagus Sylvilagus Nesolagus Oryctolagus Romerolagus Poelagus Rabbits are small mammals in the family Leporidae of the order Lagomorpha, found in several parts of the world. There are eight different genera in the family classified as rabbits, including the European rabbit (Oryctolagus cuniculus), cottontail rabbits (genus Sylvilagus; 13 species), and the Amami rabbit (Pentalagus furnessi, an endangered species on Amami Ōshima, Japan). There are many other species of rabbit, and these, along with pikas and hares, make up the order Lagomorpha. The male is called a buck and the female is a doe; a young rabbit is a kitten or kit. Habitat and range Rabbit habitats include meadows, woods, forests, grasslands, deserts and wetlands. Rabbits live in groups, and the best known species, the European rabbit, lives in underground burrows, or rabbit holes. A group of burrows is called a warren. More than half the world's rabbit population resides in North America. They are also native to southwestern Europe, Southeast Asia, Sumatra, some islands of Japan, and in parts of Africa and South America. They are not naturally found in most of Eurasia, where a number of species of hares are present. Rabbits first entered South America relatively Outdoor entrance to a rabbit burrow Rabbit 2 recently, as part of the Great American Interchange. Much of the continent has just one species of rabbit, the tapeti, while most of South America's southern cone is without rabbits. -
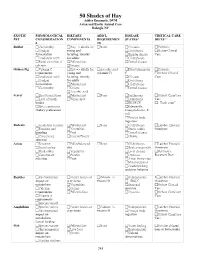
DVM, Phd, DABVP, DACVP
50 Shades of Hay Ashley Emanuele, DVM Avian and Exotic Animal Care Raleigh, NC EXOTIC PHYSIOLOGICAL DIETARY ADD’L DISEASE CRITICAL CARE PET CONSIDERATION COMPONENTS REQUIREMEN STATES* DIETS** S TS Rabbit Cecotrophy Hay => alfalfa for None GI stasis Probiotic Hindgut young and Urolithiasis Oxbow Critical fermentation lactating, timothy Bladder sludge Care Aradicular teeth for adults GI dysbiosis Renal excretion of Pelleted diet Dental disease calcium Greens Guinea Pig Vitamin C Hay => alfalfa for Ascorbic acid Hypovitaminosis Probiotic requirements young and (vitamin C) C Oxbow Critical Aradicular teeth lactating, timothy GI stasis Care Hindgut for adults Urolithiasis fermentation Pelleted diet GI dysbiosis Cecotrophy Greens Dental disease Ascorbic acid Ferret Short transit time Pelleted diet None Insulinoma Oxbow Carnivore Lack of brush Whole prey Lymphoma Care border IBD/ECE “Duck soup” Strict carnivores Salmonella, Dietary preferences Campylobacter, E. coli Foreign body ingestion Rodents Aradicular incisors Pelleted diet None GI dysbiosis Lafeber Emeraid Foraging and Vegetables Stress colitis Omnivore hoarding Fruit Dental disease Concentrate Insects/Protein Obesity selection Avian Foraging Pellet/balanced None GI dysbiosis Lafeber Emeraid Social eating diet Beak overgrowth Omnivore Beak utility Vegetables Liver disease Harrison’s Concentrate Protein Dystocia Recovery Diet selection Fruit Avian bornavirus Metal ingestion Feather picking and poor behavior Reptiles Environmental Feeder insects or Calcium +/- Endoparasites Lafeber Emeraid -

CAPYBARA (Hydrochoerus Hydrochaeris) CARE MANUAL
CAPYBARA (Hydrochoerus hydrochaeris) CARE MANUAL CREATED BY THE AZA Capybara Species Survival Plan® Program IN ASSOCIATION WITH THE AZA Rodent, Insectivore and Lagomorph TAG Capybara (Hydrochoerus hydrochaeris) Care Manual Capybara (Hydrochoerus hydrochaeris) Care Manual Published by the Association of Zoos and Aquariums in collaboration with the AZA Animal Welfare Committee. Formal Citation: AZA Rodent, Insectivore and Lagomorph TAG. (2021). Capybara Care Manual. Silver Spring, MD: Association of Zoos and Aquariums. Original Completion Date: August 2021 Authors and Significant Contributors: Sara Curtis, MPA, CVA, Akron Zoo, Research Assistant Robert Phillips, Akron Zoo, Research Assistant Shelley Orloski, CVA, Akron Zoo, Capybara Program Leader Deborah A. McWilliams, MSc, AZA-RIL TAG Nutrition, Wildlife Nutritionist Mary Agnew, PhD, Program Scientist, Wildlife Contraception Center Stephanie Chandler, Akron Zoo, Behavioral Husbandry Manager Kimberly Cook, DVM, Akron Zoo, AZA Capybara SSP Veterinary Advisor Sam Young, DVM, Greensboro Science Center, Veterinarian Monica McDonald, St Louis Zoo AZA Reproductive Management Center, Program Scientist Deniele Brown, Akron Zoo, Registrar Reviewers: Courtney Frenchak, Oak Creek Zoological Conservatory Mike Maslanka, Smithsonian National Zoological Park AZA Staff Editors: Rebecca Greenberg, Animal Programs Director Felicia Spector, (former) Animal Care Manual Editor Consultant Haley Gordon, (former) Conservation & Science Programs Assistant Hana Johnstone, (former) Conservation, Management & Welfare Sciences Intern Cover Photo Credits: Google Images Disclaimer: This manual presents a compilation of knowledge provided by recognized animal experts based on the current science, practice, and technology of animal management. The manual assembles basic requirements, best practices, and animal care recommendations to maximize capacity for excellence in animal care and welfare. The manual should be considered a work in progress, since practices continue to evolve through advances in scientific knowledge. -

Effects of Feeding Genetically Modified Cotton Seed Cake on Physiological Aspects and Meat Quality of the Rabbits (Oryctolagus Cuniculus)
Effects of Feeding Genetically Modified Cotton Seed Cake on Physiological Aspects and Meat Quality of the Rabbits (Oryctolagus cuniculus) Mubarak Abdelgabar Abdalhameed Haron B.Sc. in Science and Education, Faculty of Education, University of Zalingei (2004) Postgraduate Diploma in Biosciences and Biotechnology University of Gezira (2008) M.Sc. in Biotechnology, Faculty of Engineering and Technology University of Gezira (2010) A Thesis Submitted to the University of Gezira in Fulfillment of the Requirements for the Award of Doctor of Philosophy Degree in Biosciences and Biotechnology (Biosafety) Center of Biosciences and Biotechnology Faculty of Engineering and Technology May 2016 1 Effects of Feeding Genetically Modified Cotton Seed Cake on Physiological Aspects and Meat Quality of the Rabbits (Oryctolagus cuniculus) Mubarak Abdelgabar Abdalhameed Haron Supervision Committee Name position signature Dr. Mutaman Ali A.Kehail Main Supervisor ……........…… Prof. Elnour Elamin Abdelrahman Co-Supervisor ....……..……. Date: May 2016 2 Effects of Feeding Genetically Modified Cotton Seed Cake on Physiological Aspects and Meat Quality of the Rabbits (Oryctolagus cuniculus) Mubarak Abdelgabar Abdalhameed Haron Examination Committee Name position signature Dr. Mutaman Ali A. Kehail Chairman ………….…… Prof. Mohammed A. A. Ahmed External Examiner ………………. Prof. Mohamed Yousif Elbeeli Internal Examiner …………….… Date of Examination: 16/ 5 /2016 3 DEDICATION To the Sudanese people and Humanity with gratitude, I dedicate this work. 4 ACKNOWLEDGEMENTS Firstly, I am grateful to my God who enabled me to complete this work. With great respect and gratitude, I thank the Management of the University of Zalingei, the University of Geneina and the Ministry of High Education and Scientific Research for the opportunity of the study, I am deeply grateful to my supervisors Dr. -

Woodlands the Alberta Foundation for the Arts Travelling Exhibition Program the Interpretive Guide
Interpretive Guide & Hands-on Activities The Alberta Foundation for the Arts Travelling Exhibition Program Woodlands The Alberta Foundation for the Arts Travelling Exhibition Program The Interpretive Guide The Art Gallery of Alberta is pleased to present your community with a selection from its Travelling Exhibition Program. This is one of several exhibitions distributed by The Art Gallery of Alberta as part of the Alberta Foundation for the Arts Travelling Exhibition Program. This Interpretive Guide has been specifically designed to complement the exhibition you are now hosting. The suggested topics for discussion and accompanying activities can act as a guide to increase your viewers’ enjoyment and to assist you in developing programs to complement the exhibition. Questions and activities have been included at both elementary and advanced levels for younger and older visitors. At the Elementary School Level the Alberta Art Curriculum includes four components to provide students with a variety of experiences. These are: Reflection: Responses to visual forms in nature, designed objects and artworks Depiction: Development of imagery based on notions of realism Composition: Organization of images and their qualities in the creation of visual art Expression: Use of art materials as a vehicle for expressing statements The Secondary Level focuses on three major components of visual learning. These are: Drawings: Examining the ways we record visual information and discoveries Encounters: Meeting and responding to visual imagery Composition: Analyzing the ways images are put together to create meaning The activities in the Interpretive Guide address one or more of the above components and are generally suited for adaptation to a range of grade levels. -

Wild Bunny Rehabilitation
INTRODUCTION They come in from one day old to six weeks old, sometimes full-grown. Most of them are injured, covered with ticks, emaciated, dehydrated, whacked with a weedwacker, mowed by a lawnmower, mauled and/or degloved by a cat, or uncovered and handled by curious humans. They are the lowest mammal in Nature’s chain, and so delicate in structure as to be almost impossible to save, rehabilitate and release. They are a “pure” animal, insofar as they eat only plant life. Therefore, they represent to the many who caretake them the ultimate in vulnerability and innocence. This manual is dedicated to the thousand and then some cottontails that have been touched, healed, nursed, released and buried by these hands. They have taught me acceptance, release, faith, a deep belief in miracles and so much more. Their total dependence and acceptance of us as caregivers is a true gift. It is because of this gift that I offer to you, other rabbit caregivers, this book of experience and knowledge. Mine is not the only way, and this manual is offered as a source of information only. May you see many lightbulbs go “on”, may you see more success than you thought you could ever accomplish, and may the flightless wonders in your care return to you time and again to teach and finish the life lessons they bring to us. This manual is dedicated to all rabbit caregivers and to our sleepy-eyed wonders. There are two special sections, Feeding Tips and Feeding and Handling Techniques that were graciously written for me by: Elaine Peterson Long Texas and Federal Permits TWRC, IWRC, NWRA Rehabbing 4 years: wild rabbits, songbirds and tree squirrels Email: [email protected] All rights reserved.