Nitrous Acid and Nitrite in the Atmosphere
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) United States Patent (10) Patent No.: US 8,569,369 B2 Kramer Et Al
USOO856.9369B2 (12) United States Patent (10) Patent No.: US 8,569,369 B2 Kramer et al. (45) Date of Patent: *Oct. 29, 2013 (54) AMINO ACID COMPOUNDS OTHER PUBLICATIONS (75) Inventors: Ronald Kramer, Phoenix, AZ (US); Jablecka et al., MedSci Monit 10(I):CR29-32 (2004). Alexander Nikolaidis, New Kallikratia Maynard et al., J. Nutr. 131:287-290 (2001). (GR) Ruel et al., J. Thorac Cardiovasc. Surg 135:762-77, 2008. Rytlewski et al., European Journal of Obstetrics & Gynecology and (73) Assignee: Thermolife International, LLC, Reproductive Biology 138:23-28 (2008). Phoenix, AZ (US) Schwedheim et al., Br J Clin Pharmacol 65(1):51-59 (2007). Smith et al., J. Thorac Cardiovasc. Surg 132:58-65 (2006). (*) Notice: Subject to any disclaimer, the term of this Rytlewski et al., Eur J. Clin Invest 35 (1):32-37 (2005). patent is extended or adjusted under 35 Ming et al., Circulation 110:3708-3714 (2004). U.S.C. 154(b) by 0 days. Romero et al., Cardiovascular Drug Reviews 24(3-4):275-290 (2006). This patent is Subject to a terminal dis Oka et al., Vasc Med 10:265-274 (2005). claimer. Hayashi et al., PNAS 102(38): 13681-13686 (2005). Grasemann et al., Eur Respir J 25:62-68 (2005). (21) Appl. No.: 13/468,231 Boger, J. Nutr 137:1650S-1655S (2007). Beghetti et al., J. Thorac Cardiovasc. Surg 132(6): 1501-1502 (2006). (22) Filed: May 10, 2012 Larsen et al., B. Acta Physiol 191(1):59-66 (2007). Berge, Journal of Pharmaceutical Sciences, Jan. 1977, vol. -

Guidance for Identification and Naming of Substance Under REACH
Guidance for identification and naming of substances under 3 REACH and CLP Version 2.1 - May 2017 GUIDANCE Guidance for identification and naming of substances under REACH and CLP May 2017 Version 2.1 2 Guidance for identification and naming of substances under REACH and CLP Version 2.1 - May 2017 LEGAL NOTICE This document aims to assist users in complying with their obligations under the REACH and CLP regulations. However, users are reminded that the text of the REACH and CLP Regulations is the only authentic legal reference and that the information in this document does not constitute legal advice. Usage of the information remains under the sole responsibility of the user. The European Chemicals Agency does not accept any liability with regard to the use that may be made of the information contained in this document. Guidance for identification and naming of substances under REACH and CLP Reference: ECHA-16-B-37.1-EN Cat. Number: ED-07-18-147-EN-N ISBN: 978-92-9495-711-5 DOI: 10.2823/538683 Publ.date: May 2017 Language: EN © European Chemicals Agency, 2017 If you have any comments in relation to this document please send them (indicating the document reference, issue date, chapter and/or page of the document to which your comment refers) using the Guidance feedback form. The feedback form can be accessed via the EVHA Guidance website or directly via the following link: https://comments.echa.europa.eu/comments_cms/FeedbackGuidance.aspx European Chemicals Agency Mailing address: P.O. Box 400, FI-00121 Helsinki, Finland Visiting address: Annankatu 18, Helsinki, Finland Guidance for identification and naming of substances under 3 REACH and CLP Version 2.1 - May 2017 PREFACE This document describes how to name and identify a substance under REACH and CLP. -

WL-13 Safety Data Sheet, Canada (SDS-WL-13-CA
WL- 13 Safety Data Sheet according to the Hazardous Products Regulation (February 11, 2015) SECTION 1: Identification 1.1. Product identifier Product form : Mixture Product name : WL- 13 1.2. Recommended use and restrictions on use No additional information available 1.3. Supplier Swagelok Supplier: 29495 F.A. Lennon Drive Distributor, add your contact information 44139 Solon, OH - United States T 440-349-5600 - F 440-519-3304 www.swagelok.com 1.4. Emergency telephone number Emergency number : Infotrac: North America: 1-800-535-5053 International: 1-352-323-3500 SECTION 2: Hazard identification 2.1. Classification of the substance or mixture Classification (GHS-CA) Not classified 2.2. GHS Label elements, including precautionary statements GHS-CA labeling No labeling applicable 2.3. Other hazards No additional information available 2.4. Unknown acute toxicity (GHS-CA) No data available SECTION 3: Composition/Information on ingredients 3.1. Substances Not applicable 3.2. Mixtures Name Chemical name / Synonyms Product identifier % Classification (GHS-CA) Molybdenum(IV) sulfide Molybdenum disulfide / Molybdenum (CAS-No.) 1317-33-5 <= 10 Comb. Dust disulphide / Molybdenum sulfide / Molybdenum sulfide (MoS2) / Manganese tetroxide Sodium nitrite Diazotizing salts / Nitrous acid, (CAS-No.) 7632-00-0 <= 1.8 Acute Tox. 3 (Oral), H301 sodium salt / Nitrous acid, sodium Aquatic Acute 1, H400 salt (1:1) / SODIUM NITRITE Full text of hazard classes and H-statements: see section 16 SECTION 4: First-aid measures 4.1. Description of first aid measures First-aid measures after inhalation : Allow victim to breathe fresh air. Allow the victim to rest. First-aid measures after skin contact : Remove affected clothing and wash all exposed skin area with mild soap and water, followed by warm water rinse. -

Acids and Bases
Name Date Class CHAPTER 14 REVIEW Acids and Bases SECTION 1 SHORT ANSWER Answer the following questions in the space provided. 1. Name the following compounds as acids: sulfuric acid a. H2SO4 sulfurous acid b. H2SO3 hydrosulfuric acid c. H2S perchloric acid d. HClO4 hydrocyanic acid e. hydrogen cyanide 2. H2S Which (if any) of the acids mentioned in item 1 are binary acids? 3. Write formulas for the following acids: HNO2 a. nitrous acid HBr b. hydrobromic acid H3PO4 c. phosphoric acid CH3COOH d. acetic acid HClO e. hypochlorous acid 4. Calcium selenate has the formula CaSeO4. H2SeO4 a. What is the formula for selenic acid? H2SeO3 b. What is the formula for selenous acid? 5. Use an activity series to identify two metals that will not generate hydrogen gas when treated with an acid. Choose from Cu, Ag, Au, Pt, Pd, or Hg. 6. Write balanced chemical equations for the following reactions of acids and bases: a. aluminum metal with dilute nitric acid ϩ → ϩ 2Al(s) 6HNO3(aq) 2Al(NO3)3(aq) 3H2(g) b. calcium hydroxide solution with acetic acid ϩ → ϩ Ca(OH)2(aq) 2CH3COOH(aq) Ca(CH3COO)2(aq) 2H2O(l ) MODERN CHEMISTRY ACIDS AND BASES 117 Copyright © by Holt, Rinehart and Winston. All rights reserved. Name Date Class SECTION 1 continued 7. Write net ionic equations that represent the following reactions: a. the ionization of HClO3 in water ϩ → ϩ ϩ Ϫ HClO3(aq) H2O(l ) H3O (aq) ClO3 (aq) b. NH3 functioning as an Arrhenius base ϩ → ϩ ϩ Ϫ NH3(aq) H2O(l ) ← NH4 (aq) OH (aq) 8. -

Mechanisms of Nitric Oxide Reactions Mediated by Biologically Relevant Metal Centers
Struct Bond (2014) 154: 99–136 DOI: 10.1007/430_2013_117 # Springer-Verlag Berlin Heidelberg 2013 Published online: 5 October 2013 Mechanisms of Nitric Oxide Reactions Mediated by Biologically Relevant Metal Centers Peter C. Ford, Jose Clayston Melo Pereira, and Katrina M. Miranda Abstract Here, we present an overview of mechanisms relevant to the formation and several key reactions of nitric oxide (nitrogen monoxide) complexes with biologically relevant metal centers. The focus will be largely on iron and copper complexes. We will discuss the applications of both thermal and photochemical methodologies for investigating such reactions quantitatively. Keywords Copper Á Heme models Á Hemes Á Iron Á Metalloproteins Á Nitric oxide Contents 1 Introduction .................................................................................. 101 2 Metal-Nitrosyl Bonding ..................................................................... 101 3 How Does the Coordinated Nitrosyl Affect the Metal Center? .. .. .. .. .. .. .. .. .. .. .. 104 4 The Formation and Decay of Metal Nitrosyls ............................................. 107 4.1 Some General Considerations ........................................................ 107 4.2 Rates of NO Reactions with Hemes and Heme Models ............................. 110 4.3 Mechanistic Studies of NO “On” and “Off” Reactions with Hemes and Heme Models ................................................................................. 115 4.4 Non-Heme Iron Complexes .......................................................... -

Amino-Nitrogen Contents of Wool and Collagen
U. S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS RESEARCH PAPER RP787 Part of Journal of Research of the National Bureau of Standards, Volume 14, May 1935 AMINO-NITROGEN CONTENTS OF WOOL AND COLLAGEN By Joseph R. Kanagy 1 and Milton Harris 2 ABSTRACT When wool, collagen, and arginine were treated with nitrous acid, increasing amounts of nitrogen were evolved with time. The continued evolution of nitro gen was due to the action of nitrous acid on the guanidine nuclei of these materials. A new method for the determination of the arginine content of a protein is given. The method is based on the relative rates of evolution of nitrogen from the guanidine nuclei in a protein and in arginine. Evidence is presented t o show that the action of nitrous acid on the guanidine nucleus is different from its action on a free amino group. The free amino-nitro gen contents of wool and collagen were calculated by subtracting from the total nitrogen evolved that portion of nitrogen which came from the guanidine nuclei. The values obtained for the percentages of the total nitrogen as amino nitrogen are 2.53 for wool and 2.77 for collagen. CONTENTS Page I. Introduction ___ _____________ __________________________________ _ 563 II. Materials and methods _____________________________________ ____ _ 564 III. 566 IV. 569 Conclu~on _____________________________ __ _____________________ _ V. ~frc~~~~~~~~~~======================= ===== ========= =========== 573 VI. References __ _____ ___________ _____ ___________________ _______ ___ _ 573 I. INTRODUCTION The free amino groups in wool and collagen have been related to the combination of these materials with acids, dyes, and tannins by various investigators. -
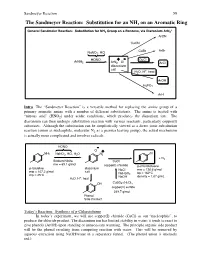
The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring
Sandmeyer Reaction 59 The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring + General Sandmeier Reaction: Substitution for NH2 Group on a Benzene, via Diazonium ArN2 ArCN CuCN CuBr ArBr NaNO2, HCl HONO + - CuCl ArNH2 ArN2 Cl ArCl diazonium salt + H2O, H , heat ArOH H3PO2 ArH Intro The “Sandmeyer Reaction” is a versatile method for replacing the amine group of a primary aromatic amine with a number of different substitutents. The amine is treated with “nitrous acid” (HNO2) under acidic conditions, which produces the diazonium ion. The diazonium can then undergo substitution reaction with various reactants, particularly copper(I) substrates. Although the substitution can be simplistically viewed as a direct ionic substitution reaction (anion as nucleophile, molecular N2 as a premier leaving group), the actual mechanism is actually more complicated and involves radicals. HONO - Cl + NH2 NaNO2, HCl, H2O N2 Cl + N2 Sodium Nitrite CuCl mw = 69.1 g/mol copper(I) chloride p-chlorotoluene p-toluidine diazonium NaCl mw = 126.6 g/mol mw = 107.2 g/mol salt NaHSO bp = 162ºC mp = 45ºC 3 + NaOH density = 1.07 g/mL H2O, H , heat OH CuSO4-(H2O)5 copper(II) sulfate 249.7 g/mol Phenol Side Product Today’s Reaction: Synthesis of p-Chlorotoluene In today’s experiment, we will use copper(I) chloride (CuCl) as our “nucleophile”, to produce the chloride product. The diazonium ion has limited stability in water; it tends to react to give phenols (ArOH) upon standing or unnecessary warming. The principle organic side product will be the phenol resulting from competing reaction with water. -

Different Nitrous Acid Sources in the Air Quality Levels
Discussion Paper | Discussion Paper | Discussion Paper | Discussion Paper | Atmos. Chem. Phys. Discuss., 10, 28183–28230, 2010 Atmospheric www.atmos-chem-phys-discuss.net/10/28183/2010/ Chemistry ACPD doi:10.5194/acpd-10-28183-2010 and Physics 10, 28183–28230, 2010 © Author(s) 2010. CC Attribution 3.0 License. Discussions Different nitrous acid This discussion paper is/has been under review for the journal Atmospheric Chemistry sources in the air and Physics (ACP). Please refer to the corresponding final paper in ACP if available. quality levels The impact of different nitrous acid M. Gonc¸alves et al. sources in the air quality levels of the Title Page Iberian Peninsula Abstract Introduction Conclusions References M. Gonc¸alves1,2, D. Dabdub3, W. L. Chang3, F. Saiz3, O. Jorba2, and J. M. Baldasano1,2 Tables Figures 1Environmental Modeling Laboratory, Projects Department, Technical University of Catalonia, Barcelona, Spain J I 2Earth Sciences Department, Barcelona Supercomputing Center, Barcelona, Spain J I 3Department of Mechanical and Aerospace Engineering, University of California, Irvine, Irvine, California, USA Back Close Received: 21 October 2010 – Accepted: 5 November 2010 – Published: 17 November 2010 Full Screen / Esc Correspondence to: M. Gonc¸alves ([email protected]) Printer-friendly Version Published by Copernicus Publications on behalf of the European Geosciences Union. Interactive Discussion 28183 Discussion Paper | Discussion Paper | Discussion Paper | Discussion Paper | Abstract ACPD Hydroxyl radical (OH) is a primary oxidant in the atmosphere and affects both gas- phase pollutants and particulate matter levels. Nitrous acid (HONO) acts as an impor- 10, 28183–28230, 2010 tant source of OH in the urban atmosphere. -

Preparation of Hydroxy Analogues of Several Amino Acids
PRE PARA TION OF HYDROXY ANA LOGUES OF SE\~RA L AMI NO ACIDS by THOMAS LE E ABRAHAMS A THESIS submitted to OREGON STA TE COLLEGE in partial fulfillment of the requirements for the degree of MAS TER OF SCIE NCE June 1957 APPROIIED t Redacted for Privacy Profcasor of Chcmlstry In Chsrgc of, !{aJsr lnd Heed of Chculatry Redacted for Privacy Chalrnen of $choo1 Oraduate 0onn!,ttor Redacted for Privacy Dren of 0mduata Sohool Date thoar.c ta prcso"t"a 7, / t y,fl,,Zf^*? fyptd by Barbsra Jr l{ol,coa ACK NOWLEDGMENT The author wishes to express his gratitude to Dr. Bert E. Christensen for his guidance and encouragement throughout the course of this research. TABLE OF CONTENTS Introduction .,.., -- ....... ... ... .. 1 Experimental .... - ... - - - 14 Summary~ __ ~ ~ ~ ~ ~ __ ........... - 21 Bibliography _____ - - .. - .. .. - 23 PREPARATION OF HYDROXY ANALOGUES OF SEVERAL AMINO ACIDS In 1911, Van Slyke (28, pp .l92-202} reported a procedure for the analysis of proteins by the determina tion of amino groups which are characteristic of most amino acids. Essentially , the method was adapted from the earlier Hausmann method (13, pp~l3S-145) as modified by Osborne and Harris (17, pp.,332-353) with his own meth od for the determination of amino nitrogen. In his orig inal study dealing with the method, Van Slyke (28, pp .l92 202) showed that the majority of the naturally occurring amino acids with the exception of glycine and cystine~ when treated with nitrous acid, yield their amino nitro gen quantitatively at room temperature. More recently, -

Nitrite and Nitrosyl Compounds in Food Preservation
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1411 (1999) 475^488 Review Nitrite and nitrosyl compounds in food preservation Richard Cammack a;*, C.L. Joannou 1;a, Xiao-Yuan Cui 2;a, Claudia Torres Martinez 3;b, Shaun R. Maraj b, Martin N. Hughes b a Division of Life Sciences, King's College, London W8 7AH, UK b Department of Chemistry, King's College, London WC2R 2LS, UK Received 27 August 1998; received in revised form 2 November 1998; accepted 16 December 1998 Abstract Nitrite is consumed in the diet, through vegetables and drinking water. It is also added to meat products as a preservative. The potential risks of this practice are balanced against the unique protective effect against toxin-forming bacteria such as Clostridium botulinum. The chemistry of nitrite, and compounds derived from it, in food systems and bacterial cells are complex. It is known that the bactericidal species is not nitrite itself, but a compound or compounds derived from it during 3 food preparation. Of a range of nitrosyl compounds tested, the anion of Roussin's black salt [Fe4S3(NO)7] was the most inhibitory to C. sporogenes. This compound is active against both anaerobic and aerobic food-spoilage bacteria, while some other compounds are selective, indicating multiple sites of action. There are numerous possible targets for inhibition in the bacterial cells, including respiratory chains, iron^sulfur proteins and other metalloproteins, membranes and the genetic apparatus. ß 1999 Elsevier Science B.V. All rights reserved. Keywords: Roussin's salts; Nitrosothiol; Electron paramagnetic resonance spectroscopy; (Clostridium botulinum); (Listeria monocytogenes) Contents 1. -

Acids and Bases Acids Release H+(Aq) When Dissolved in Water and Bases Release HO-(Aq)
Acids and Bases Acids release H+(aq) when dissolved in water and bases release HO-(aq). Strong acids and bases dissociate completely in water. Weak acids and bases are in equilibrium with H+(aq) and HO- (aq), respectively. Acids react with bases to form water in neutralization reactions. Outline • Strong Acids and Bases • Weak Acids • Homework Litmus paper for pH determination Strong Acids and Bases Strong Acids in Water Strong acids are those that completely dissociate in water. You've seen them all in a previous section: HCl, HBr, HI, H2SO4, HNO3, and HClO4. Chemistry 102 Prof. Shapley page 1 The concentration of solvated protons is equal to the moles of the acid added to each liter of water. When 0.05 mol of HNO3 is added to 50 mL water: [H+] = (0.05 mol)/(50 mL)(1 L/1000 mL) = 1.0 M pH = -log(1.0) = -log(100) = 0 When 0.05 mol of H2SO4 is added to 10 L water: [H+] = (0.05 mol)/(10 L) = 0.005 M = 5 x 10-3 = 100.70 x 10-3 pH = -log(10-2.3) = 2.3 Strong Bases in Water The strongest base that can exist in water is HO-. There are many hydroxide salts that provide HO- when dissolved in water. These include NaOH, KOH, Mg(OH)2, Ca(OH)2, and Al(OH)3. There are other bases that are even stronger than hydroxide but these react with water to make hydroxide. [Li+][NH2-] + H2O NH3 + Li+ + HO- [Na+][H-] + H2O H2(g) + Na+ + HO- Neutralization Reactions The reaction between a strong acid and an equal number of moles of a strong base produces water. -

Nitrogen Oxides (Nox), Why and How They Are Controlled
United States Office of Air Quality EPA 456/F-99-006R Environmental Protection Planning and Standards November 1999 Agency Research Triangle Park, NC 27711 Air EPA-456/F-99-006R November 1999 Nitrogen Oxides (NOx), Why and How They Are Controlled Prepared by Clean Air Technology Center (MD-12) Information Transfer and Program Integration Division Office of Air Quality Planning and Standards U.S. Environmental Protection Agency Research Triangle Park, North Carolina 27711 DISCLAIMER This report has been reviewed by the Information Transfer and Program Integration Division of the Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency and approved for publication. Approval does not signify that the contents of this report reflect the views and policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products is not intended to constitute endorsement or recommendation for use. Copies of this report are available form the National Technical Information Service, U.S. Department of Commerce, 5285 Port Royal Road, Springfield, Virginia 22161, telephone number (800) 553-6847. CORRECTION NOTICE This document, EPA-456/F-99-006a, corrects errors found in the original document, EPA-456/F-99-006. These corrections are: Page 8, fourth paragraph: “Destruction or Recovery Efficiency” has been changed to “Destruction or Removal Efficiency;” Page 10, Method 2. Reducing Residence Time: This section has been rewritten to correct for an ambiguity in the original text. Page 20, Table 4. Added Selective Non-Catalytic Reduction (SNCR) to the table and added acronyms for other technologies. Page 29, last paragraph: This paragraph has been rewritten to correct an error in stating the configuration of a typical cogeneration facility.