CCM2 Molecular Signaling Pathway Arianne J
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Detection of Aneuploidies by Paralogous Sequence Quantification S Deutsch, U Choudhury, G Merla, C Howald, a Sylvan, S E Antonarakis
908 J Med Genet: first published as 10.1136/jmg.2004.023184 on 9 December 2004. Downloaded from ORIGINAL ARTICLE Detection of aneuploidies by paralogous sequence quantification S Deutsch, U Choudhury, G Merla, C Howald, A Sylvan, S E Antonarakis ............................................................................................................................... J Med Genet 2004;41:908–915. doi: 10.1136/jmg.2004.023184 Background: Chromosomal aneuploidies are a common cause of congenital disorders associated with cognitive impairment and multiple dysmorphic features. Pre-natal diagnosis of aneuploidies is most See end of article for commonly performed by the karyotyping of fetal cells obtained by amniocentesis or chorionic villus authors’ affiliations sampling, but this method is labour intensive and requires about 14 days to complete. ....................... Methods: We have developed a PCR based method for the detection of targeted chromosome number Correspondence to: abnormalities termed paralogous sequence quantification (PSQ), based on the use of paralogous genes. Professor Stylianos E Paralogous sequences have a high degree of sequence identity, but accumulate nucleotide substitutions in Antonarakis, Department a locus specific manner. These sequence differences, which we term paralogous sequence mismatches of Genetic Medicine and Development, University of (PSMs), can be quantified using pyrosequencing technology, to estimate the relative dosage between Geneva Medical School, different chromosomes. We designed 10 assays for the detection of trisomies of chromosomes 13, 18, and GE 1211, Geneva, 21 and sex chromosome aneuploidies. Switzerland; Stylianos. antonarakis@medecine. Results: We evaluated the performance of this method on 175 DNAs, highly enriched for abnormal unige.ch samples. A correct and unambiguous diagnosis was given for 119 out of 120 aneuploid samples as well as for all the controls. -

Novel Mutation and Three Other Sequence Variants Segregating with Phenotype at Keratoconus 13Q32 Susceptibility Locus
European Journal of Human Genetics (2012) 20, 389–397 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus Marta Czugala1,6, Justyna A Karolak1,6, Dorota M Nowak1, Piotr Polakowski2, Jose Pitarque3, Andrea Molinari3, Malgorzata Rydzanicz1, Bassem A Bejjani4, Beatrice YJT Yue5, Jacek P Szaflik2 and Marzena Gajecka*,1 Keratoconus (KTCN), a non-inflammatory corneal disorder characterized by stromal thinning, represents a major cause of corneal transplantations. Genetic and environmental factors have a role in the etiology of this complex disease. Previously reported linkage analysis revealed that chromosomal region 13q32 is likely to contain causative gene(s) for familial KTCN. Consequently, we have chosen eight positional candidate genes in this region: MBNL1, IPO5, FARP1, RNF113B, STK24, DOCK9, ZIC5 and ZIC2, and sequenced all of them in 51 individuals from Ecuadorian KTCN families and 105 matching controls. The mutation screening identified one mutation and three sequence variants showing 100% segregation under a dominant model with KTCN phenotype in one large Ecuadorian family. These substitutions were found in three different genes: c.2262A4C (p.Gln754His) and c.720+43A4GinDOCK9; c.2377-132A4CinIPO5 and c.1053+29G4CinSTK24. PolyPhen analyses predicted that c.2262A4C (Gln754His) is possibly damaging for the protein function and structure. Our results suggest that c.2262A4C (p.Gln754His) -

Identification of Cis-Regulatory Sequence
Worsley-Hunt et al. Genome Medicine 2011, 3:65 http://genomemedicine.com/content/3/9/65 REVIEW Identication of cis-regulatory sequence variations in individual genome sequences Rebecca Worsley-Hunt1,2, Virginie Bernard1 and Wyeth W Wasserman1* Abstract for the appropriate patterning or magnitude of gene expression. Similarly, mutations can disrupt other sequence- Functional contributions of cis-regulatory sequence specific regulatory controls, such as elements regulating variations to human genetic disease are numerous. For RNA splicing or stability. Although much emphasis in the instance, disrupting variations in a HNF4A transcription age of exome sequencing has been placed on variation factor binding site upstream of the Factor IX gene within protein-encoding sequences, it is apparent that contributes causally to hemophilia B Leyden. Although regulatory sequence disruptions will become a key focus clinical genome sequence analysis currently focuses as full genome sequences become widely accessible for on the identication of protein-altering variation, the medical genetics research. impact of cis-regulatory mutations can be similarly e emerging collection of cis-regulatory variations strong. New technologies are now enabling genome that cause human disease or altered phenotype is grow- sequencing beyond exomes, revealing variation across ing [2-4]. Reports identify cis-acting, expression-altering the non-coding 98% of the genome responsible for mutations observed within introns, far upstream of developmental and physiological patterns of gene genes, at splicing sites or within microRNA target sites. activity. The capacity to identify causal regulatory For example, cis-regulatory mutations have roles in hemo- mutations is improving, but predicting functional philia, Gilbert’s syndrome, Bernard-Soulier syndrome, changes in regulatory DNA sequences remains a great irritable bowel syndrome, beta-thalassemia, cholesterol challenge. -

Transcriptome-Wide Profiling of Cerebral Cavernous Malformations
www.nature.com/scientificreports OPEN Transcriptome-wide Profling of Cerebral Cavernous Malformations Patients Reveal Important Long noncoding RNA molecular signatures Santhilal Subhash 2,8, Norman Kalmbach3, Florian Wegner4, Susanne Petri4, Torsten Glomb5, Oliver Dittrich-Breiholz5, Caiquan Huang1, Kiran Kumar Bali6, Wolfram S. Kunz7, Amir Samii1, Helmut Bertalanfy1, Chandrasekhar Kanduri2* & Souvik Kar1,8* Cerebral cavernous malformations (CCMs) are low-fow vascular malformations in the brain associated with recurrent hemorrhage and seizures. The current treatment of CCMs relies solely on surgical intervention. Henceforth, alternative non-invasive therapies are urgently needed to help prevent subsequent hemorrhagic episodes. Long non-coding RNAs (lncRNAs) belong to the class of non-coding RNAs and are known to regulate gene transcription and involved in chromatin remodeling via various mechanism. Despite accumulating evidence demonstrating the role of lncRNAs in cerebrovascular disorders, their identifcation in CCMs pathology remains unknown. The objective of the current study was to identify lncRNAs associated with CCMs pathogenesis using patient cohorts having 10 CCM patients and 4 controls from brain. Executing next generation sequencing, we performed whole transcriptome sequencing (RNA-seq) analysis and identifed 1,967 lncRNAs and 4,928 protein coding genes (PCGs) to be diferentially expressed in CCMs patients. Among these, we selected top 6 diferentially expressed lncRNAs each having signifcant correlative expression with more than 100 diferentially expressed PCGs. The diferential expression status of the top lncRNAs, SMIM25 and LBX2-AS1 in CCMs was further confrmed by qRT-PCR analysis. Additionally, gene set enrichment analysis of correlated PCGs revealed critical pathways related to vascular signaling and important biological processes relevant to CCMs pathophysiology. -
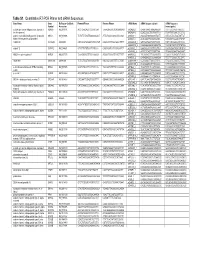
Table S1. Quantitative RT-PCR Primer and Sirna Sequences Gene Name
Table S1. Quantitative RT-PCR Primer and siRNA Sequences Gene Name Gene RefSeq or GenBank Forward Primer Reverse Primer siRNA Name siRNA Sequence (plus) siRNA Sequence Symbol Accession (minus/guide) a disintegrin and metalloproteinase domain 9 ADAM9 NM_003816 ACCTCAGCAGTTCCCATCAA TAAAGGAGGTGCAGGAGCAG siADAM9_1 GAGATTAACTAGAGAAAGA TCTTTCTCTAGTTAATCTC (meltrin gamma) siADAM9_2 GGAGGGAGTTCATAATTCA TGAATTATGAACTCCCTCC guanine nucleotide binding protein (G protein), GNAI3 NM_006496 TCTGTTATAGTTGGCGGCAGT ATTCTGCAAAGGCAAAAGGA siGNAI3_1 AGAACTAGCAGGAGTGATT AATCACTCCTGCTAGTTCT alpha inhibiting activity polypeptide 3 siGNAI3_2 ACACAGGTTCCAATACATA TATGTATTGGAACCTGTGT AA099748 AA099748 AA099748 CTACCACTGAGGTGTCCCTGT CAGCCATCTAACAGCATTTTT siAA099748_1 GTTAGATGGCTGATTAATA TATTAATCAGCCATCTAAC siAA099748_2 CGACAGAGGAGCAGCATTA TAATGCTGCTCCTCTGTCG calpain 12 CAPN12 NM_144691 GTCCTTCTGTCCCTCATCCA CAGCAGCTCCTCTGGAATCT siCAPN12_1 GGACACGCGTATTCCATCA TGATGGAATACGCGTGTCC siCAPN12_2 AATCCTCAGTTCCGTTTAA TTAAACGGAACTGAGGATT NMDA receptor regulated 1 NARG1 NM_057175 TAAAGGGAATTTGCCGAAGA AGCACTGGAGTTCGGTTTGT siNARG1_1 TGCGAGATCTTGAGGGTTA TAACCCTCAAGATCTCGCA siNARG1_2 GATAGGAGGTCCAAAAGAA TTCTTTTGGACCTCCTATC AK091308 AK091308 AK091308 TCTCCTGAATGGGAGGAATG GGCAACAGATGTTTCCTGTG siAK091308_1 CCAAAGACTTAGACTGTAA TTACAGTCTAAGTCTTTGG siAK091308_2 GCACAAAGCATTTACAACA TGTTGTAAATGCTTTGTGC serine/threonine kinase 24 (STE20 homolog, STK24 NM_003576 GCATCTGCCTTCCTTATCCA TGACAGTGTTTTGCCAGAGG siSTK24_1 TCGATTATCTCCATTCGGA TCCGAATGGAGATAATCGA yeast) siSTK24_2 CATCGGACTTGGACAGAAA -

Mir-17-92 Fine-Tunes MYC Expression and Function to Ensure
ARTICLE Received 31 Mar 2015 | Accepted 22 Sep 2015 | Published 10 Nov 2015 DOI: 10.1038/ncomms9725 OPEN miR-17-92 fine-tunes MYC expression and function to ensure optimal B cell lymphoma growth Marija Mihailovich1, Michael Bremang1, Valeria Spadotto1, Daniele Musiani1, Elena Vitale1, Gabriele Varano2,w, Federico Zambelli3, Francesco M. Mancuso1,w, David A. Cairns1,w, Giulio Pavesi3, Stefano Casola2 & Tiziana Bonaldi1 The synergism between c-MYC and miR-17-19b, a truncated version of the miR-17-92 cluster, is well-documented during tumor initiation. However, little is known about miR-17-19b function in established cancers. Here we investigate the role of miR-17-19b in c-MYC-driven lymphomas by integrating SILAC-based quantitative proteomics, transcriptomics and 30 untranslated region (UTR) analysis upon miR-17-19b overexpression. We identify over one hundred miR-17-19b targets, of which 40% are co-regulated by c-MYC. Downregulation of a new miR-17/20 target, checkpoint kinase 2 (Chek2), increases the recruitment of HuR to c- MYC transcripts, resulting in the inhibition of c-MYC translation and thus interfering with in vivo tumor growth. Hence, in established lymphomas, miR-17-19b fine-tunes c-MYC activity through a tight control of its function and expression, ultimately ensuring cancer cell homeostasis. Our data highlight the plasticity of miRNA function, reflecting changes in the mRNA landscape and 30 UTR shortening at different stages of tumorigenesis. 1 Department of Experimental Oncology, European Institute of Oncology, Via Adamello 16, Milan 20139, Italy. 2 Units of Genetics of B cells and lymphomas, IFOM, FIRC Institute of Molecular Oncology Foundation, Milan 20139, Italy. -

CCM1 and CCM2 Variants in Patients with Cerebral Cavernous
www.nature.com/scientificreports OPEN CCM1 and CCM2 variants in patients with cerebral cavernous malformation in an ethnically Received: 19 January 2018 Accepted: 11 July 2019 Chinese population in Taiwan Published: xx xx xxxx Chun-Wei Chang 1, Peng-Wei Hsu2,3, Kuo-Chen Wei2,3, Chia-Wen Chang1, Hon-Chung Fung1, Mo-Song Hsih1, Wen-Chuin Hsu1,3, Long-Sun Ro1,3, Chen-Nen Chang2,4, Jiun-Jie Wang5,6, Yih-Ru Wu1,3 & Sien-Tsong Chen1 Cerebral cavernous malformation (CCM) is a vascular malformation characterized by clustered enlarged capillary-like channels in the central nervous system. The genes harboring variants in patients with CCM include CCM1/Krev interaction trapped-1, CCM2/MGC4607, and CCM3/programmed cell death protein 10. We aimed to identify pathogenic variants in an ethnic Chinese population in Taiwan. We recruited 95 patients with multiple CCMs or a single lesion with a relevant family history. Sanger sequencing was performed for 41 patients. Variants were identifed using sequence alignment tools, and the clinical signifcance of these variants was determined using American College of Medical Genetics and Genomics standards and guidelines. Several pathogenic variants were found in six patients, including three unrelated patients and three afected members of one family. Two novel pathogenic variants leading to early truncation comprised a deletion variant in exon 18 of CCM1 (c.1846delA; p.Glu617LysfsTer44) and an insertion variant in exon 4 of CCM2 (c.401_402insGCCC; p.Ile136AlafsTer4). One novel pathogenic splice site variant was c.485 + 1G > C at the beginning of intron 8 of CCM1. In this study, we identifed novel variants related to CCM in an ethnically Chinese population in Taiwan. -

STK24 Antibody - C-Terminal Region Rabbit Polyclonal Antibody Catalog # AI16132
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 STK24 Antibody - C-terminal region Rabbit Polyclonal Antibody Catalog # AI16132 Specification STK24 Antibody - C-terminal region - Product Information Application WB Primary Accession Q9Y6E0 Other Accession NP_003567 Reactivity Human Host Rabbit Clonality Polyclonal Calculated MW 48kDa KDa STK24 Antibody - C-terminal region - Additional Information Host: Rabbit Target Name: STK24 Gene ID 8428 Sample Tissue: 293T Whole Cell lysates Antibody Dilution: 1.0μg/ml Alias Symbol STK24, MST3, STK3, Other Names STK24 Antibody - C-terminal region - Serine/threonine-protein kinase 24, Background 2.7.11.1, Mammalian STE20-like protein kinase 3, MST-3, STE20-like kinase MST3, Serine/threonine-protein kinase that acts on Serine/threonine-protein kinase 24 36 kDa both serine and threonine residues and subunit, Mammalian STE20-like protein promotes apoptosis in response to stress kinase 3 N-terminal, MST3/N, stimuli and caspase activation. Mediates Serine/threonine-protein kinase 24 12 kDa oxidative-stress- induced cell death by subunit, Mammalian STE20-like protein modulating phosphorylation of JNK1-JNK2 kinase 3 C-terminal, MST3/C, STK24, MST3, (MAPK8 and MAPK9), p38 (MAPK11, MAPK12, STK3 MAPK13 and MAPK14) during oxidative stress. Plays a role in a staurosporine-induced Format caspase- independent apoptotic pathway by Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and regulating the nuclear translocation of AIFM1 2% sucrose. and ENDOG and the DNase activity associated with ENDOG. Phosphorylates STK38L on Reconstitution & Storage 'Thr-442' and stimulates its kinase activity. Add 50 &mu, l of distilled water. -

MST3 (STK24) (NM 001032296) Human Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC209117L4V MST3 (STK24) (NM_001032296) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: MST3 (STK24) (NM_001032296) Human Tagged ORF Clone Lentiviral Particle Symbol: STK24 Synonyms: HEL-S-95; MST3; MST3B; STE20; STK3 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) ACCN: NM_001032296 ORF Size: 1293 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC209117). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_001032296.2 RefSeq Size: 4624 bp RefSeq ORF: 1296 bp Locus ID: 8428 UniProt ID: Q9Y6E0, Q5U0E6, Q6P0Y1 Protein Families: Druggable Genome, Protein Kinase MW: 47.7 kDa Gene Summary: This gene encodes a serine/threonine protein kinase that functions upstream of mitogen- activated protein kinase (MAPK) signaling. The encoded protein is cleaved into two chains by caspases; the N-terminal fragment (MST3/N) translocates to the nucleus and promotes programmed cells death. -

Cerebral Cavernous Malformations: from Genes to Proteins to Disease
See the corresponding editorial in this issue, pp 119–121. J Neurosurg 116:122–132, 2012 Cerebral cavernous malformations: from genes to proteins to disease Clinical article DANIEL D. CAVALCANTI, M.D., M. YASHAR S. KALANI, M.D., PH.D., NIKOLAY L. MARTIROSYAN, M.D., JUSTIN EALES, B.S., ROBERT F. SPETZLER, M.D., AND MARK C. PREUL, M.D. Division of Neurological Surgery, Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center, Phoenix, Arizona Over the past half century molecular biology has led to great advances in our understanding of angio- and vas- culogenesis and in the treatment of malformations resulting from these processes gone awry. Given their sporadic and familial distribution, their developmental and pathological link to capillary telangiectasias, and their observed chromosomal abnormalities, cerebral cavernous malformations (CCMs) are regarded as akin to cancerous growths. Although the exact pathological mechanisms involved in the formation of CCMs are still not well understood, the identification of 3 genetic loci has begun to shed light on key developmental pathways involved in CCM pathogen- esis. Cavernous malformations can occur sporadically or in an autosomal dominant fashion. Familial forms of CCMs have been attributed to mutations at 3 different loci implicated in regulating important processes such as proliferation and differentiation of angiogenic precursors and members of the apoptotic machinery. These processes are important for the generation, maintenance, and pruning of every vessel in the body. In this review the authors highlight the lat- est discoveries pertaining to the molecular genetics of CCMs, highlighting potential new therapeutic targets for the treatment of these lesions. -

STRIPAK Complexes in Cell Signaling and Cancer
Oncogene (2016), 1–9 © 2016 Macmillan Publishers Limited All rights reserved 0950-9232/16 www.nature.com/onc REVIEW STRIPAK complexes in cell signaling and cancer Z Shi1,2, S Jiao1 and Z Zhou1,3 Striatin-interacting phosphatase and kinase (STRIPAK) complexes are striatin-centered multicomponent supramolecular structures containing both kinases and phosphatases. STRIPAK complexes are evolutionarily conserved and have critical roles in protein (de) phosphorylation. Recent studies indicate that STRIPAK complexes are emerging mediators and regulators of multiple vital signaling pathways including Hippo, MAPK (mitogen-activated protein kinase), nuclear receptor and cytoskeleton remodeling. Different types of STRIPAK complexes are extensively involved in a variety of fundamental biological processes ranging from cell growth, differentiation, proliferation and apoptosis to metabolism, immune regulation and tumorigenesis. Growing evidence correlates dysregulation of STRIPAK complexes with human diseases including cancer. In this review, we summarize the current understanding of the assembly and functions of STRIPAK complexes, with a special focus on cell signaling and cancer. Oncogene advance online publication, 15 February 2016; doi:10.1038/onc.2016.9 INTRODUCTION in the central nervous system and STRN4 is mostly abundant in Recent proteomic studies identified a group of novel multi- the brain and lung, whereas STRN3 is ubiquitously expressed in 5–9 component complexes named striatin (STRN)-interacting phos- almost all tissues. STRNs share a -

A Focus on Genetic Features of Cerebral Cavernous Malformations and Brain Arteriovenous Malformations Pathogenesis
Neurological Sciences (2019) 40:243–251 https://doi.org/10.1007/s10072-018-3674-x REVIEW ARTICLE Vis-à-vis: a focus on genetic features of cerebral cavernous malformations and brain arteriovenous malformations pathogenesis Concetta Scimone1,2 & Luigi Donato 1,2 & Silvia Marino3 & Concetta Alafaci1 & Rosalia D’Angelo1 & Antonina Sidoti1,2 Received: 7 September 2018 /Accepted: 1 December 2018 /Published online: 6 December 2018 # Fondazione Società Italiana di Neurologia 2018 Abstract Cerebrovascular malformations include a wide range of blood vessel disorders affecting brain vasculature. Neuroimaging differential diagnosis can result unspecific due to similar phenotypes of lesions and their deep locali- zation. Next-generation sequencing (NGS) platforms simultaneously analyze several hundreds of genes and can be applied for molecular distinction of different phenotypes within the same disorder’s macro-area. We discuss about the main criticisms regarding molecular bases of cerebral cavernous malformations (CCM) and brain arteriovenous malformations (AVM), highlighting both common pathogenic aspects and genetic differences leading to lesion devel- opment. Many recent studies performed on human CCM and AVM tissues aim to detect genetic markers to better understand molecular bases and pathogenic mechanism, particularly for sporadic cases. Several genes involved in angiogenesis show different expression patterns between CCM and AVM, and these could represent a valid starting point to project a NGS panel to apply for differential cerebrovascular malformation diagnosis. Keywords Cerebral cavernous malformations . Arteriovenous malformations . Genetics . Differential molecular diagnosis Introduction #116860) and arteriovenous malformations (AVM, OMIM #108010) are the most frequent, affecting more Cerebrovascular malformations include a wide spectrum than 3% of the population [1]. Venous developmental of intracranial blood vessel disorders, involving arterial anomalies (VDAs) are rare conditions usually associated wall, capillary bed, and venous and lymphatic systems.