A Generalized Method for Fissile Material Characterization Using Short-Lived Fission Product Gamma Spectroscopy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
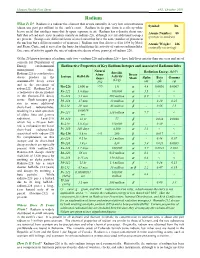
Radium What Is It? Radium Is a Radioactive Element That Occurs Naturally in Very Low Concentrations Symbol: Ra (About One Part Per Trillion) in the Earth’S Crust
Human Health Fact Sheet ANL, October 2001 Radium What Is It? Radium is a radioactive element that occurs naturally in very low concentrations Symbol: Ra (about one part per trillion) in the earth’s crust. Radium in its pure form is a silvery-white heavy metal that oxidizes immediately upon exposure to air. Radium has a density about one- Atomic Number: 88 half that of lead and exists in nature mainly as radium-226, although several additional isotopes (protons in nucleus) are present. (Isotopes are different forms of an element that have the same number of protons in the nucleus but a different number of neutrons.) Radium was first discovered in 1898 by Marie Atomic Weight: 226 and Pierre Curie, and it served as the basis for identifying the activity of various radionuclides. (naturally occurring) One curie of activity equals the rate of radioactive decay of one gram (g) of radium-226. Of the 25 known isotopes of radium, only two – radium-226 and radium-228 – have half-lives greater than one year and are of concern for Department of Energy environmental Radioactive Properties of Key Radium Isotopes and Associated Radionuclides management sites. Natural Specific Radiation Energy (MeV) Radium-226 is a radioactive Abun- Decay Isotope Half-Life Activity decay product in the dance Mode Alpha Beta Gamma (Ci/g) uranium-238 decay series (%) (α) (β) (γ) and is the precursor of Ra-226 1,600 yr >99 1.0 α 4.8 0.0036 0.0067 radon-222. Radium-228 is a radioactive decay product Rn-222 3.8 days 160,000 α 5.5 < < in the thorium-232 decay Po-218 3.1 min 290 million α 6.0 < < series. -

Table 2.Iii.1. Fissionable Isotopes1
FISSIONABLE ISOTOPES Charles P. Blair Last revised: 2012 “While several isotopes are theoretically fissionable, RANNSAD defines fissionable isotopes as either uranium-233 or 235; plutonium 238, 239, 240, 241, or 242, or Americium-241. See, Ackerman, Asal, Bale, Blair and Rethemeyer, Anatomizing Radiological and Nuclear Non-State Adversaries: Identifying the Adversary, p. 99-101, footnote #10, TABLE 2.III.1. FISSIONABLE ISOTOPES1 Isotope Availability Possible Fission Bare Critical Weapon-types mass2 Uranium-233 MEDIUM: DOE reportedly stores Gun-type or implosion-type 15 kg more than one metric ton of U- 233.3 Uranium-235 HIGH: As of 2007, 1700 metric Gun-type or implosion-type 50 kg tons of HEU existed globally, in both civilian and military stocks.4 Plutonium- HIGH: A separated global stock of Implosion 10 kg 238 plutonium, both civilian and military, of over 500 tons.5 Implosion 10 kg Plutonium- Produced in military and civilian 239 reactor fuels. Typically, reactor Plutonium- grade plutonium (RGP) consists Implosion 40 kg 240 of roughly 60 percent plutonium- Plutonium- 239, 25 percent plutonium-240, Implosion 10-13 kg nine percent plutonium-241, five 241 percent plutonium-242 and one Plutonium- percent plutonium-2386 (these Implosion 89 -100 kg 242 percentages are influenced by how long the fuel is irradiated in the reactor).7 1 This table is drawn, in part, from Charles P. Blair, “Jihadists and Nuclear Weapons,” in Gary A. Ackerman and Jeremy Tamsett, ed., Jihadists and Weapons of Mass Destruction: A Growing Threat (New York: Taylor and Francis, 2009), pp. 196-197. See also, David Albright N 2 “Bare critical mass” refers to the absence of an initiator or a reflector. -

Combating Illicit Trafficking in Nuclear and Other Radioactive Material Radioactive Other Traffickingand Illicit Nuclear Combating in 6 No
8.8 mm IAEA Nuclear Security Series No. 6 Technical Guidance Reference Manual IAEA Nuclear Security Series No. 6 in Combating Nuclear Illicit and Trafficking other Radioactive Material Combating Illicit Trafficking in Nuclear and other Radioactive Material This publication is intended for individuals and organizations that may be called upon to deal with the detection of and response to criminal or unauthorized acts involving nuclear or other radioactive material. It will also be useful for legislators, law enforcement agencies, government officials, technical experts, lawyers, diplomats and users of nuclear technology. In addition, the manual emphasizes the international initiatives for improving the security of nuclear and other radioactive material, and considers a variety of elements that are recognized as being essential for dealing with incidents of criminal or unauthorized acts involving such material. Jointly sponsored by the EUROPOL WCO INTERNATIONAL ATOMIC ENERGY AGENCY VIENNA ISBN 978–92–0–109807–8 ISSN 1816–9317 07-45231_P1309_CovI+IV.indd 1 2008-01-16 16:03:26 COMBATING ILLICIT TRAFFICKING IN NUCLEAR AND OTHER RADIOACTIVE MATERIAL REFERENCE MANUAL The Agency’s Statute was approved on 23 October 1956 by the Conference on the Statute of the IAEA held at United Nations Headquarters, New York; it entered into force on 29 July 1957. The Headquarters of the Agency are situated in Vienna. Its principal objective is “to accelerate and enlarge the contribution of atomic energy to peace, health and prosperity throughout the world’’. IAEA NUCLEAR SECURITY SERIES No. 6 TECHNICAL GUIDANCE COMBATING ILLICIT TRAFFICKING IN NUCLEAR AND OTHER RADIOACTIVE MATERIAL REFERENCE MANUAL JOINTLY SPONSORED BY THE EUROPEAN POLICE OFFICE, INTERNATIONAL ATOMIC ENERGY AGENCY, INTERNATIONAL POLICE ORGANIZATION, AND WORLD CUSTOMS ORGANIZATION INTERNATIONAL ATOMIC ENERGY AGENCY VIENNA, 2007 COPYRIGHT NOTICE All IAEA scientific and technical publications are protected by the terms of the Universal Copyright Convention as adopted in 1952 (Berne) and as revised in 1972 (Paris). -

The Use of Cosmic-Rays in Detecting Illicit Nuclear Materials
The use of Cosmic-Rays in Detecting Illicit Nuclear Materials Timothy Benjamin Blackwell Department of Physics and Astronomy University of Sheffield This dissertation is submitted for the degree of Doctor of Philosophy 05/05/2015 Declaration I hereby declare that except where specific reference is made to the work of others, the contents of this dissertation are original and have not been submitted in whole or in part for consideration for any other degree or qualification in this, or any other Univer- sity. This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration, except where specifically indicated in the text. Timothy Benjamin Blackwell 05/05/2015 Acknowledgements This thesis could not have been completed without the tremendous support of many people. Firstly I would like to express special appreciation and thanks to my academic supervisor, Dr Vitaly A. Kudryavtsev for his expertise, understanding and encourage- ment. I have enjoyed our many discussions concerning my research topic throughout the PhD journey. I would also like to thank my viva examiners, Professor Lee Thomp- son and Dr Chris Steer, for the time you have taken out of your schedules, so that I may take this next step in my career. Thanks must also be given to Professor Francis Liven, Professor Neil Hyatt and the rest of the Nuclear FiRST DTC team, for the initial oppor- tunity to pursue postgraduate research. Appreciation is also given to the University of Sheffield, the HEP group and EPRSC for providing me with the facilities and funding, during this work. -

ESTIMATION of FISSION-PRODUCT GAS PRESSURE in URANIUM DIOXIDE CERAMIC FUEL ELEMENTS by Wuzter A
NASA TECHNICAL NOTE NASA TN D-4823 - - .- j (2. -1 "-0 -5 M 0-- N t+=$j oo w- P LOAN COPY: RET rm 3 d z c 4 c/) 4 z ESTIMATION OF FISSION-PRODUCT GAS PRESSURE IN URANIUM DIOXIDE CERAMIC FUEL ELEMENTS by WuZter A. PuuZson una Roy H. Springborn Lewis Reseurcb Center Clevelund, Ohio NATIONAL AERONAUTICS AND SPACE ADMINISTRATION WASHINGTON, D. C. NOVEMBER 1968 i 1 TECH LIBRARY KAFB, NM I 111111 lllll IllH llll lilll1111111111111 Ill1 01317Lb NASA TN D-4823 ESTIMATION OF FISSION-PRODUCT GAS PRESSURE IN URANIUM DIOXIDE CERAMIC FUEL ELEMENTS By Walter A. Paulson and Roy H. Springborn Lewis Research Center Cleveland, Ohio NATIONAL AERONAUTICS AND SPACE ADMINISTRATION For sale by the Clearinghouse for Federal Scientific and Technical Information Springfield, Virginia 22151 - CFSTl price $3.00 ABSTRACl Fission-product gas pressure in macroscopic voids was calculated over the tempera- ture range of 1000 to 2500 K for clad uranium dioxide fuel elements operating in a fast neutron spectrum. The calculated fission-product yields for uranium-233 and uranium- 235 used in the pressure calculations were based on experimental data compiled from various sources. The contributions of cesium, rubidium, and other condensible fission products are included with those of the gases xenon and krypton. At low temperatures, xenon and krypton are the major contributors to the total pressure. At high tempera- tures, however, cesium and rubidium can make a considerable contribution to the total pressure. ii ESTIMATION OF FISSION-PRODUCT GAS PRESSURE IN URANIUM DIOXIDE CERAMIC FUEL ELEMENTS by Walter A. Paulson and Roy H. -

A Fissile Material Cut-Off Treaty N I T E D Understanding the Critical Issues N A
U N I D I R A F i s s i l e M a A mandate to negotiate a treaty banning the production of fissile material t e r i for nuclear weapons has been under discussion in the Conference of a l Disarmament (CD) in Geneva since 1994. On 29 May 2009 the Conference C u on Disarmament agreed a mandate to begin those negotiations. Shortly t - o afterwards, UNIDIR, with the support of the Government of Switzerland, f f T launched a project to support this process. r e a t This publication is a compilation of various products of the project, y : that hopefully will help to illuminate the critical issues that will need to U n be addressed in the negotiation of a treaty that stands to make a vital d e r contribution to the cause of nuclear disarmament and non-proliferation. s t a n d i n g t h e C r i t i c a l I s s u e s UNITED NATIONS INSTITUTE FOR DISARMAMENT RESEARCH U A Fissile Material Cut-off Treaty N I T E D Understanding the Critical Issues N A Designed and printed by the Publishing Service, United Nations, Geneva T I GE.10-00850 – April 2010 – 2,400 O N UNIDIR/2010/4 S UNIDIR/2010/4 A Fissile Material Cut-off Treaty Understanding the Critical Issues UNIDIR United Nations Institute for Disarmament Research Geneva, Switzerland New York and Geneva, 2010 Cover image courtesy of the Offi ce of Environmental Management, US Department of Energy. -

Compilation and Evaluation of Fission Yield Nuclear Data Iaea, Vienna, 2000 Iaea-Tecdoc-1168 Issn 1011–4289
IAEA-TECDOC-1168 Compilation and evaluation of fission yield nuclear data Final report of a co-ordinated research project 1991–1996 December 2000 The originating Section of this publication in the IAEA was: Nuclear Data Section International Atomic Energy Agency Wagramer Strasse 5 P.O. Box 100 A-1400 Vienna, Austria COMPILATION AND EVALUATION OF FISSION YIELD NUCLEAR DATA IAEA, VIENNA, 2000 IAEA-TECDOC-1168 ISSN 1011–4289 © IAEA, 2000 Printed by the IAEA in Austria December 2000 FOREWORD Fission product yields are required at several stages of the nuclear fuel cycle and are therefore included in all large international data files for reactor calculations and related applications. Such files are maintained and disseminated by the Nuclear Data Section of the IAEA as a member of an international data centres network. Users of these data are from the fields of reactor design and operation, waste management and nuclear materials safeguards, all of which are essential parts of the IAEA programme. In the 1980s, the number of measured fission yields increased so drastically that the manpower available for evaluating them to meet specific user needs was insufficient. To cope with this task, it was concluded in several meetings on fission product nuclear data, some of them convened by the IAEA, that international co-operation was required, and an IAEA co-ordinated research project (CRP) was recommended. This recommendation was endorsed by the International Nuclear Data Committee, an advisory body for the nuclear data programme of the IAEA. As a consequence, the CRP on the Compilation and Evaluation of Fission Yield Nuclear Data was initiated in 1991, after its scope, objectives and tasks had been defined by a preparatory meeting. -

Nuclear Power
No.59 z iii "Ill ~ 2 er0 Ill Ill 0 Nuclear Family Pia nning p3 Chernobyl Broadsheet ·, _ I. _ . ~~~~ George Pritchar d speaks CONTENTS COMMENT The important nuclear development since the Nuclear Family Planning 3 last SCRAM Journal was the Government's The CEGB's plans, and the growing opposition, after Sizewell B by go ahead for Sizewell B: the world's first HUGH RICHARDS. reactor order since Chernobyl, and Britain's News 4-6 first since the go ahead was given to Torness Accidents Will Happen 1 and Heysham 2 in 1978. Of great concern is Hinkley Seismic Shocker 8-9 the CEGB's announced intention to build "a A major article on seismic safety of nuclear plants in which JAMES small fanilty• of PWRs, starting with Hinkley GARRETT reveals that Hinkley Point C. At the time of the campaign In the Point sits on a geological fault. south west to close the Hinkley A Magnox Trouble at Trawsfynydd 10-11 station, and .a concerted push in Scotland to A summary of FoE's recent report on increasing radiation levels from prevent the opening of Torness, another Trawsfynydd's by PATRICK GREEN. nuclear announcement is designed to divide Pandora's POX 12 and demoralise the opposition. But, it should The debate over plutonium transport make us more determined. The article on the to and from Dounreay continues by facing page gives us hope: the local PETE MUTTON. authorities on Severnside are joining forces CHERNOBYL BROADSHEET to oppose Hinkley C, and hopefully they will Cock-ups and Cover-ups work closely with local authorities in other "Sacrificed to • • • Nuclear Power" threatened areas - Lothian Region, The Soviet Experience Northumberland, the County Council Coalition "An Agonising Decision• 13 against waste dumping and the Nuclear Free GEORGE PRITCHARD explains why Zones - to formulate a national anti-nuclear he left Greenpeoce and took a job strategy. -

A New Methodology for Determining Fissile Mass in Individual Accounting Items with the Use of Gamma-Ray Spectrometry*
BNL-67176 A NEW METHODOLOGY FOR DETERMINING FISSILE MASS IN INDIVIDUAL ACCOUNTING ITEMS WITH THE USE OF GAMMA-RAY SPECTROMETRY* Walter R. Kane, Peter E. Vanier, Peter B. Zuhoski, and James R. Lemley Brookhaven National Laboratory Building 197C, P. O. Box 5000, Upton, NY 11973-5000 USA 631/344-3841 FAX 631/344-7533 Abstract In the safeguards, arms control, and nonproliferation regimes measurements are required which give the quantity of fissile material in an accounting item, e.g., a standard container of plutonium or uranium oxide. Because of the complexity of modeling the absorption of gamma rays in high-Z materials, gamma-ray spectrometry is not customarily used for this purpose. Gamma-ray measurements can be used to determine the fissile mass when two conditions are met: 1. The material is in a standard container, and 2. The material is finely divided, or a solid item with a reproducible shape. The methodology consists of: A. Measurement of the emitted gamma rays, and B. Measurement of the transmission through the item of the high-energy gamma rays of Co-60 and Th-228. We have demonstrated that items containing nuclear materials possess a characteristic "fingerprint" of gamma rays which depends not only on the nuclear properties, but also on the mass, density, shape, etc.. The material's spectrum confirms its integrity, homogeneity, and volume as well. While there is attenuation of radiation from the interior, the residual radiation confirms the homogeneity of the material throughout the volume. Transmission measurements, where the attenuation depends almost entirely on Compton scattering, determine the material mass. -

Relative Fission Product Yield Determination in the Usgs
RELATIVE FISSION PRODUCT YIELD DETERMINATION IN THE USGS TRIGA MARK I REACTOR by Michael A. Koehl © Copyright by Michael A. Koehl, 2016 All Rights Reserved A thesis submitted to the Faculty and the Board of Trustees of the Colorado School of Mines in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Nuclear Engineering). Golden, Colorado Date: ____________________ Signed: ________________________ Michael A. Koehl Signed: ________________________ Dr. Jenifer C. Braley Thesis Advisor Golden, Colorado Date: ____________________ Signed: ________________________ Dr. Mark P. Jensen Professor and Director Nuclear Science and Engineering Program ii ABSTRACT Fission product yield data sets are one of the most important and fundamental compilations of basic information in the nuclear industry. This data has a wide range of applications which include nuclear fuel burnup and nonproliferation safeguards. Relative fission yields constitute a major fraction of the reported yield data and reduce the number of required absolute measurements. Radiochemical separations of fission products reduce interferences, facilitate the measurement of low level radionuclides, and are instrumental in the analysis of low-yielding symmetrical fission products. It is especially useful in the measurement of the valley nuclides and those on the extreme wings of the mass yield curve, including lanthanides, where absolute yields have high errors. This overall project was conducted in three stages: characterization of the neutron flux in irradiation positions within the U.S. Geological Survey TRIGA Mark I Reactor (GSTR), determining the mass attenuation coefficients of precipitates used in radiochemical separations, and measuring the relative fission products in the GSTR. Using the Westcott convention, the Westcott flux, ; modified spectral index, ; neutron temperature, ; and gold-based cadmium ratiosφ were determined for various sampling√⁄ positions in the USGS TRIGA Mark I reactor. -

Ornl-Tm-1853
OAK RIDGE NATIONAL LABORATORY operated by UNION CARBIDE CORPORATION for the U.S. ATOMIC ENERGY COMMISSION ORNL- TM-1853 COPY NO. - 0-C kc+- DATE -6-6-67 CFSTI RpICZiS CHEMICAL RESEARCHAND DEVELOPMENTFOR MOLTEN- SALT BREEDERREACTORS W. R. Grimes ABSTRACT kq Results of the 15-year program of chemical research and develop- .; ment for molten salt reactors are summarized in this document. These c results indicate that 7LiF-BeFz-LJFb mixtures are feasible fuels for thermal breeder reactors. Such mixtures show satisfactory phase be- havior, they are compatible with Hastelloy N and moderator graphite, and they appear to resist radiation and tolerate fission product ac- cumulation. Mixtures of 7LiF-BeF2-ThF4 similarly appear suitable as blankets for such machines. Several possible secondary coolant mix- tures are available; NaF-NaBF3 systems seem, at present, to be the most likely possibility. Gaps in the technology are presented along with the accomplish- ments, and an attempt is made to define the information (and the research and development program) needed before a Molten Salt Thermal Breeder can be operated with confidence. NOTICE This document contains information of a preliminary nature and was prepared primarily for internal use at the Oak Ridge National Loboratory. It is subject to revision or correction and therefore does not represent a final report. The information is not to be abstracted, reprinted or otherwise given public dis- 4 .”: semination without the approval of the ORNL potent branch, Legal and Infor- mation Control Department. ’ y a I LEGAL NOTICE This report was prepored as an occount of Government sponsored work. Neither the United States, nw the Commission, nor ony person octing un beholf of the Commission: A. -

Two-Proton Radioactivity 2
Two-proton radioactivity Bertram Blank ‡ and Marek P loszajczak † ‡ Centre d’Etudes Nucl´eaires de Bordeaux-Gradignan - Universit´eBordeaux I - CNRS/IN2P3, Chemin du Solarium, B.P. 120, 33175 Gradignan Cedex, France † Grand Acc´el´erateur National d’Ions Lourds (GANIL), CEA/DSM-CNRS/IN2P3, BP 55027, 14076 Caen Cedex 05, France Abstract. In the first part of this review, experimental results which lead to the discovery of two-proton radioactivity are examined. Beyond two-proton emission from nuclear ground states, we also discuss experimental studies of two-proton emission from excited states populated either by nuclear β decay or by inelastic reactions. In the second part, we review the modern theory of two-proton radioactivity. An outlook to future experimental studies and theoretical developments will conclude this review. PACS numbers: 23.50.+z, 21.10.Tg, 21.60.-n, 24.10.-i Submitted to: Rep. Prog. Phys. Version: 17 December 2013 arXiv:0709.3797v2 [nucl-ex] 23 Apr 2008 Two-proton radioactivity 2 1. Introduction Atomic nuclei are made of two distinct particles, the protons and the neutrons. These nucleons constitute more than 99.95% of the mass of an atom. In order to form a stable atomic nucleus, a subtle equilibrium between the number of protons and neutrons has to be respected. This condition is fulfilled for 259 different combinations of protons and neutrons. These nuclei can be found on Earth. In addition, 26 nuclei form a quasi stable configuration, i.e. they decay with a half-life comparable or longer than the age of the Earth and are therefore still present on Earth.