Synthesis and Study of Photocatalytic and Conducting Nanoparticles And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WATER CHEMISTRY CONTINUING EDUCATION PROFESSIONAL DEVELOPMENT COURSE 1St Edition
WATER CHEMISTRY CONTINUING EDUCATION PROFESSIONAL DEVELOPMENT COURSE 1st Edition 2 Water Chemistry 1st Edition 2015 © TLC Printing and Saving Instructions The best thing to do is to download this pdf document to your computer desktop and open it with Adobe Acrobat DC reader. Adobe Acrobat DC reader is a free computer software program and you can find it at Adobe Acrobat’s website. You can complete the course by viewing the course materials on your computer or you can print it out. Once you’ve paid for the course, we’ll give you permission to print this document. Printing Instructions: If you are going to print this document, this document is designed to be printed double-sided or duplexed but can be single-sided. This course booklet does not have the assignment. Please visit our website and download the assignment also. You can obtain a printed version from TLC for an additional $69.95 plus shipping charges. All downloads are electronically tracked and monitored for security purposes. 3 Water Chemistry 1st Edition 2015 © TLC We require the final exam to be proctored. Do not solely depend on TLC’s Approval list for it may be outdated. A second certificate of completion for a second State Agency $25 processing fee. Most of our students prefer to do the assignment in Word and e-mail or fax the assignment back to us. We also teach this course in a conventional hands-on class. Call us and schedule a class today. Responsibility This course contains EPA’s federal rule requirements. Please be aware that each state implements drinking water/wastewater/safety regulations may be more stringent than EPA’s or OSHA’s regulations. -

=« Y * MASS SPECTROMETRIC STUDIES of the SYNTHESIS
"In presenting the dissertation as a partial fulfillment of the requirements for an advanced degree from the Georgia Institute of Technology, I agree that the Library of the Institution shall make it available for inspection and circulation in accordance with its regulations governing materials of this type. I agree that permission to copy from, or to publish from, this dissertation may be granted by the professor under whose direction it was written, or, in his absence, by the dean of the Graduate Division when such copying or publication is solely for scholarly purposes and does not involve potential financial gain. It is understood that any copying from, or publication of, this dissertation which involves potential financial gain will not be allowed without written permission, "-- -. • i -=« y * MASS SPECTROMETRIC STUDIES OF THE SYNTHESIS, REACTIVITY, AND ENERGETICS OF THE OXYGEN FLUORIDES AT CRYOGENIC TEMPERATURES A THESIS Presented to The Faculty of the Graduate Division by Thomas Joseph Malone In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the School of Chemical Engineering Georgia Institute of Technology February, 1966 MASS SPECTROMETRIC STUDIES OF THE SYNTHESIS, REACTIVITY, AND ENERGETICS OF THE OXYGEN FLUORIDES AT CRYOGENIC TEMPERATURES Approved; / • ' "/v Date approved by Chairma n:\PuJl 3 lUt, 11 ACKNOWLEDGMENTS I am deeply indebted to Dr0 Ho Ao McGee, Jr0 for his encouragement, advice^ and assistance throughout both my graduate and undergraduate studieso Many hours of discussion and willing -
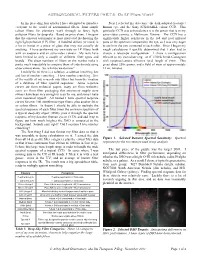
ASTRONOMICAL FILTERS PART 5: Do LP Filters Work?
ASTRONOMICAL FILTERS PART 5: Do LP Filters Work? In my preceding four articles I have attempted to introduce Next I selected my detectors: the dark adapted (scotopic) everyone to the world of astronomical filters, from simple human eye, and the Sony ICX418AKL colour CCD. This colour filters for planetary work through to fancy light particular CCD was selected since it is the sensor that is in my pollution filters for deep-sky. Based on price alone, I imagine astro-video camera, a Mallincam Xtreme. The CCD has a that the amateur astronomer is most interested in choosing the significantly higher sensitivity in the red and near infrared right light pollution (LP) filter. A hundred dollars (or more) is parts of the spectrum compared to the eye, so I was very eager a lot to invest in a piece of glass that may not actually do to see how the two compared to each other. Once I began my anything. I have performed my own tests on LP filters, both rough calculations I quickly determined that I also had to with an eyepiece and an astro-video camera. My tests have choose a telescope configuration. I chose a configuration been limited to only a couple of different filter types and relevant to my own observing: an 8” f/10 Schmidt-Cassegrain brands. The shear numbers of filters on the market make it with eyepiece/camera effective focal length of 8mm. This pretty much impossible to compare them all side-by-side using gives about 250x power, and a field of view of approximately observations alone. -

CCD Imaging of Planetary Nebulae Or H II Regions
Observational Astronomy ASTR 310 Fall 2010 Project 2 CCD Imaging of Planetary Nebulae or H II Regions 1. Introduction The aim of this project is to gain experience using the CCD camera by obtaining images of planetary nebulae or H II regions. Planetary nebulae are ionized clouds of gas surrounding very hot stars. The nebula is formed when a star of moderate mass reaches the end of its life as a red giant star, and makes the transition to its final state as a white dwarf. In this process the outer layers of the star are ejected to form the nebula, while the newly exposed core appears as a very hot central star, which emits most of its radiation in the far ultraviolet, thus ionizing and heating the nebula. Most of the radiation emitted by the nebular gas, which has a temperature of ∼10,000 K, is concentrated in the emission lines of a few elements such as oxygen and nitrogen, as well as the most abundant element, hydrogen. An H II region is an interstellar gas cloud with newly formed stars in or near it which are hot enough to ionize some of the gas. While the physical conditions in the gas and the emitted spectrum are similar to planetary nebulae, H II regions are much less regular in appearance and contain much more gas and dust. We will take the images of the nebulae through two narrow-band interference filters which transmit the light of the strongest emission lines in the nebular spectrum. These are the Hα line of hydrogen at 656.3 nm and the line of doubly ionized oxygen (O++ or [O III]) at 500.7 nm. -

Enrichment of the Interstellar Medium by Metal-Rich Droplets and the Abundance Bias in H Ii Regions
A&A 471, 193–204 (2007) Astronomy DOI: 10.1051/0004-6361:20065675 & c ESO 2007 Astrophysics Enrichment of the interstellar medium by metal-rich droplets and the abundance bias in H ii regions G. Stasinska´ 1, G. Tenorio-Tagle2, M. Rodríguez2, and W. J. Henney3 1 LUTH, Observatoire de Paris, CNRS, Université Paris Diderot, Place Jules Janssen, 92190 Meudon, France e-mail: [email protected] 2 Instituto Nacional de Astrofísica Óptica y Electrónica, AP 51, 72000 Puebla, Mexico 3 Centro de Radioastronomía y Astrofísica, Universidad Nacional Autónoma de México, Campus Morelia, Apartado Postal 3-72, 58090 Morelia, Mexico Received 23 May 2006 / Accepted 25 May 2007 ABSTRACT We critically examine a scenario for the enrichment of the interstellar medium (ISM) in which supernova ejecta follow a long (108 yr) journey before falling back onto the galactic disk in the form of metal-rich “droplets”, These droplets do not become fully mixed with the interstellar medium until they become photoionized in H ii regions. We investigate the hypothesis that the photoionization of these highly metallic droplets can explain the observed “abundance discrepancy factors” (ADFs), which are found when comparing abundances derived from recombination lines and from collisionally excited lines, both in Galactic and extragalactic H ii regions. We derive bounds of 1013–1015 cm on the droplet sizes inside H ii regions in order that (1) they should not have already been detected by direct imaging of nearby nebulae, and (2) they should not be too swiftly destroyed by diffusion in the ionized gas. From photoionization modelling we find that, if this inhomogeneous enrichment scenario holds, then the recombination lines strongly overestimate the metallicities of the fully mixed H ii regions. -

Amateur Astronomy Filters
Amateur Astronomy Filters Eyepiece filters usually screw into the bottom of the eyepiece and come in sizes that fit both 1.25 inch and 2 inch eyepieces. These filters can enhance the contrast of low surface brightness objects like diffuse nebula. They can be used to en- hance the surface features of the planets and Moon, reduce glare of very bright A N T E L O P E V A L L E Y objects, and even enhance the definition of images. There are filters that reduce ASTRONOMY CLUB, INC. the effects of light pollution and those that allow the spectrum to pass through at A 501 (c)(3) Non-Profit only one frequency. Several are summarized below. Organization Solar Filter: This is the filter that will allow you to safely observe the closest star On the web at: to the Earth - the Sun. These filters fit snugly over the front of the telescope and www.avastronomyclub.org are made out of either coated glass, or a Mylar material. Blue Filter: A Wratten #44A, 47B or 80A is used to detect high altitude clouds on Mars, white ovals and spots in the belts of Jupiter, and the zones of the clouds of Saturn, and to reduce the glare of the bright Moon. The 80A is the filter to have if Tel: (661) 724-1623 you only buy one filter. VALLEY ANTELOPE Or (661) 822-4580 Green Filter: A Wratten #58 allows you to see more clearly the edges of the Martian polar caps and enhances the belts and the Great Red Spot in the clouds INC. -

A Second Flavor of Wolf-Rayet Central Stars of Planetary Nebulae
by Brent Miszalski, Paul Crowther, and Anthony Moffat A Second Flavor of Wolf-Rayet Central Stars of Planetary Nebulae Gemini South optical observations of the planetary nebula IC 4663 reveal the first proven case of a central star with a nitrogen-sequence Wolf- Rayet spectrum. Its existence challenges the conventional view of how certain solar-mass stars become hydrogen-deficient white dwarfs. Population I classical Wolf-Rayet stars represent the short-lived, hydrogen-deficient, pre-su- pernova phase of very massive stars. These hot, high-luminosity bodies possess powerful, fast, dense winds (for recent review see Crowther, 2007). They exhibit unique, broad emission lines generated via Doppler expansion that are readily seen spectroscopically. Wolf-Rayet stars come in two main flavors: nitrogen-rich WN-type stars and carbon-rich WC- type stars. These two classes reflect the products of hydrogen and helium-burning, respec- tively; whereas helium and nitrogen emission lines dominate in WN-types, the WC-types show emission lines mostly of carbon, oxygen, and helium. Very high-mass stars are thought to end their lives as WN or WC stars, although they are exceptionally rare, with only a few hundred cases known in the Milky Way. The Wolf-Rayet Star Phenomenon A subset of low-mass, post-Asymptotic Giant Branch (AGB) stars are also hydrogen-deficient (Werner Herwig, 2006). High temperature examples include He-rich subdwarf OB stars, O(He) stars, and DO white dwarfs. In addition, around 100 hydrogen-deficient central stars of plan- etary nebulae also exhibit a Wolf-Rayet spectroscopic signature. To date, all Wolf-Rayet central stars have been carbon-rich variants, with square brackets added to distinguish [WC]-type central stars from WC stars. -

Oxygen Vacancies and Lithium Vacancies
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Faculty Publications, Department of Physics and Astronomy Research Papers in Physics and Astronomy 2010 Identification of electron and hole traps in lithium tetraborate (Li2B4O7) crystals: Oxygen vacancies and lithium vacancies M. W. Swinney J. W. McClory J. C. Petrosky Shan Yang (??) A. T. Brant See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/physicsfacpub This Article is brought to you for free and open access by the Research Papers in Physics and Astronomy at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Faculty Publications, Department of Physics and Astronomy by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors M. W. Swinney, J. W. McClory, J. C. Petrosky, Shan Yang (??), A. T. Brant, V. T. Adamiv, Ya. V. Burak, P. A. Dowben, and L. E. Halliburton JOURNAL OF APPLIED PHYSICS 107, 113715 ͑2010͒ Identification of electron and hole traps in lithium tetraborate „Li2B4O7… crystals: Oxygen vacancies and lithium vacancies ͒ M. W. Swinney,1 J. W. McClory,1,a J. C. Petrosky,1 Shan Yang (杨山͒,2 A. T. Brant,2 V. T. Adamiv,3 Ya. V. Burak,3 P. A. Dowben,4 and L. E. Halliburton2 1Department of Engineering Physics, Air Force Institute of Technology, Wright-Patterson Air Force Base, Ohio 45433, USA 2Department of Physics, West Virginia University, Morgantown, West Virginia 26506, USA 3Institute of Physical Optics, Dragomanov 23, L’viv 79005, Ukraine 4Department of Physics and Astronomy, Nebraska Center for Materials and Nanoscience, University of Nebraska, Lincoln, Nebraska 68588, USA ͑Received 7 January 2010; accepted 18 March 2010; published online 11 June 2010͒ Electron paramagnetic resonance ͑EPR͒ and electron-nuclear double resonance ͑ENDOR͒ are used to identify and characterize electrons trapped by oxygen vacancies and holes trapped by lithium ͑ ͒ vacancies in lithium tetraborate Li2B4O7 crystals. -
![Arxiv:1611.08305V1 [Astro-Ph.GA] 24 Nov 2016](https://docslib.b-cdn.net/cover/2024/arxiv-1611-08305v1-astro-ph-ga-24-nov-2016-6822024.webp)
Arxiv:1611.08305V1 [Astro-Ph.GA] 24 Nov 2016
Draft version November 28, 2016 Typeset using LATEX twocolumn style in AASTeX61 NEBULAR CONTINUUM AND LINE EMISSION IN STELLAR POPULATION SYNTHESIS MODELS Nell Byler,1 Julianne J. Dalcanton,1 Charlie Conroy,2 and Benjamin D. Johnson2 1Department of Astronomy, University of Washington, Box 351580, Seattle, WA 98195, USA 2Department of Astronomy, Harvard University, Cambridge, MA, USA (Received November 16, 2016) Submitted to ApJ ABSTRACT Accounting for nebular emission when modeling galaxy spectral energy distributions (SEDs) is important, as both line and continuum emission can contribute significantly to the total observed flux. In this work, we present a new nebular emission model integrated within the Flexible Stellar Population Synthesis code that computes the total line and continuum emission for complex stellar populations using the photoionization code Cloudy. The self-consistent coupling of the nebular emission to the matched ionizing spectrum produces emission line intensities that correctly scale with the stellar population as a function of age and metallicity. This more complete model of galaxy SEDs will improve estimates of global gas properties derived with diagnostic diagrams, star formation rates based on Hα, and stellar masses derived from NIR broadband photometry. Our models agree well with results from other photoionization models and are able to reproduce observed emission from H ii regions and star-forming galaxies. Our models show improved agreement with the observed H ii regions in the Ne iii/O ii plane and show satisfactory agreement with He ii emission from z = 2 galaxies when including rotating stellar models. Models including post-asymptotic giant branch stars are able to reproduce line ratios consistent with low-ionization emission regions (LIERs). -

An Analytic Model for OIII Fine Structure Emission from High Redshift Galaxies
MNRAS 000,1–16 (2018) Preprint 30 July 2020 Compiled using MNRAS LATEX style file v3.0 An analytic model for [O iii] fine structure emission from high redshift galaxies Shengqi Yang,1? Adam Lidz2 1Center for Cosmology and Particle Physics, Department of physics, New York University, 726 Broadway, New York, NY, 10003, U.S.A. 2Department of Physics and Astronomy, University of Pennsylvania, 209 South 33rd Street, Philadelphia, PA 19104, USA Accepted XXX. Received YYY; in original form ZZZ ABSTRACT Recent ALMA measurements have revealed bright [O iii] 88 micron line emission from galax- ies during the Epoch of Reionization (EoR) at redshifts as large as z ∼ 9. We introduce an analytic model to help interpret these and other upcoming [O iii] 88 micron measurements. Our approach sums over the emission from discrete Str¨omgren spheres and considers the total volume of ionized hydrogen in a galaxy of a given star-formation rate. We estimate the relative volume of doubly-ionized oxygen and ionized hydrogen and its dependence on the spectrum of ionizing photons. We then calculate the level populations of OIII ions in different fine-structure states for HII regions of specified parameters. In this simple model, a galaxy’s [O iii] 88 µm lu- minosity is determined by: the typical number density of free electrons in HII regions (ne), the average metallicity of these regions (Z), the rate of hydrogen ionizing photons emitted (QHI), and the shape of the ionizing spectrum. We cross-check our model by comparing it with de- tailed CLOUDY calculations, and find that it works to better than 15% accuracy across a broad range of parameter space. -

Planetary Nebulae
Spectroscopy of Gaseous Nebulae in Galaxies M. J. Barlow, Dept of Physics & Astronomy, CL O TLINE: (i) Overview of ionized nebulae: HII regions, planetary nebulae and supernova remnants (ii) Determining nebular temperatures, densities and ionic abundances from collisionally excited lines (iii) Measuring element abundances in external galaxies (iv) Probing nebulae with deep spectra: s.process element forbidden lines and recombination lines from C, N, O and Ne. (v) Heavy element abundances from forbidden lines vs. those from optical recombination lines Photoioni)ed Nebulae: two main types HII regions: the brightest examples are the result of the formation of massive stars (> 20 Msun), which then ionize and disperse the natal cloud. The ionizing stars have temperatures of 2 ,000 K " Tstar " 0,000 K. Planetary Nebulae: the final stage in the life of low and intermediate mass stars before they become white dwarfs. The outer envelope is ejected during the red giant ,-. phase and then photoionized by the hot remnant star, with 2 ,000 K " Tstar " 200,000 K. The Orion Nebula (M40) ,n /00 region blister at the front of the 1rion Molecular 2loud Tstar 6 38,000K Large 9uantities of dust can be present in /00 regions 5 b ILL 5 I LL I 5 h b a #$ % D J K L JJ M % N J$ J# OL J LL J a J a JPP J , starburst galaxy with many /00 regions VLT image of NGC 1020. /00 regions show up as white in this image. Spectroscopic studies of heavy element forbidden lines have enabled element abundance gradients to be measured as a function of galactocentric distance for many galaxies. -

Stellar Atmospheres
5 a f/rr ». I lfaf?A5^‘'y )»} STELLAR ATMOSPHERES Donated by Mrs. Yemuna Bappn to The Indian Institute of Astrophysics from tbe personal collection of Dr. M. K. V. Bappu HARVARD OBSERVATORY MONOGRAPHS HARLOW SHAPLEY, Editor No. 1 STELLAR ATMOSPHERES A CONTRIBUTION TO THE OBSERVATIONAL STUDY OF HIGH TEMPERATURE IN THE REVERSING LAYERS OF STARS BY CECILIA H. PAYNE PUBLISHED BY THE OBSERVATORY CAMBRIDGE, MASSACHUSETTS 1925 ilA l-ils.. COPYRIGHT, 1925 BY HARVARD OBSERVATORY PRINTED AT THE HARVARD UNIVERSITY PRESS CAMBRIDGE, MASS., U.S.A. EDITOR’S FOREWORD The most effective way of publishing the results of astronomical investigations is clearly dependent on the nature and scope of each particular research. The Harvard Observatory has used various forms. Nearly a hundred volumes of Annals contain, for the most part, tabular material presenting observational results on the positions, photometry, and spectroscopy of stars, nebulae, and planets. Shorter investigations have been reported in Circulars, Bulletins, and in current scientific journals from which Reprints are obtained and issued serially. It now appears that a few extensive investigations of a some- what monographic nature can be most conveniently presented as books, the first of which is the present special analysis of stellar spectra by Miss Payne. Other volumes in this series, it is hoped, will be issued during the next few years, each dealing with a sub- ject in which a large amount of original investigation is being carried on at this observatory. The Monographs will differ in another respect from all the pub- lications previously issued from the Harvard Observatory — they cannot be distributed gratis to observatories and other interested scientific institutions.