Nephronophthisis, a Pediatric Challenge CASE REPORT
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Educational Paper Ciliopathies
Eur J Pediatr (2012) 171:1285–1300 DOI 10.1007/s00431-011-1553-z REVIEW Educational paper Ciliopathies Carsten Bergmann Received: 11 June 2011 /Accepted: 3 August 2011 /Published online: 7 September 2011 # The Author(s) 2011. This article is published with open access at Springerlink.com Abstract Cilia are antenna-like organelles found on the (NPHP) . Ivemark syndrome . Meckel syndrome (MKS) . surface of most cells. They transduce molecular signals Joubert syndrome (JBTS) . Bardet–Biedl syndrome (BBS) . and facilitate interactions between cells and their Alstrom syndrome . Short-rib polydactyly syndromes . environment. Ciliary dysfunction has been shown to Jeune syndrome (ATD) . Ellis-van Crefeld syndrome (EVC) . underlie a broad range of overlapping, clinically and Sensenbrenner syndrome . Primary ciliary dyskinesia genetically heterogeneous phenotypes, collectively (Kartagener syndrome) . von Hippel-Lindau (VHL) . termed ciliopathies. Literally, all organs can be affected. Tuberous sclerosis (TSC) . Oligogenic inheritance . Modifier. Frequent cilia-related manifestations are (poly)cystic Mutational load kidney disease, retinal degeneration, situs inversus, cardiac defects, polydactyly, other skeletal abnormalities, and defects of the central and peripheral nervous Introduction system, occurring either isolated or as part of syn- dromes. Characterization of ciliopathies and the decisive Defective cellular organelles such as mitochondria, perox- role of primary cilia in signal transduction and cell isomes, and lysosomes are well-known -

Unraveling the Genetics of Joubert and Meckel-Gruber Syndromes
Journal of Pediatric Genetics 3 (2014) 65–78 65 DOI 10.3233/PGE-14090 IOS Press Unraveling the genetics of Joubert and Meckel-Gruber syndromes Katarzyna Szymanska, Verity L. Hartill and Colin A. Johnson∗ Department of Ophthalmology and Neuroscience, University of Leeds, Leeds, UK Received 27 May 2014 Revised 11 July 2014 Accepted 14 July 2014 Abstract. Joubert syndrome (JBTS) and Meckel-Gruber syndrome (MKS) are recessive neurodevelopmental conditions caused by mutations in proteins that are structural or functional components of the primary cilium. In this review, we provide an overview of their clinical diagnosis, management and molecular genetics. Both have variable phenotypes, extreme genetic heterogeneity, and display allelism both with each other and other ciliopathies. Recent advances in genetic technology have significantly improved diagnosis and clinical management of ciliopathy patients, with the delineation of some general genotype-phenotype correlations. We highlight those that are most relevant for clinical practice, including the correlation between TMEM67 mutations and the JBTS variant phenotype of COACH syndrome. The subcellular localization of the known MKS and JBTS proteins is now well-described, and we discuss some of the contemporary ideas about ciliopathy disease pathogenesis. Most JBTS and MKS proteins localize to a discrete ciliary compartment called the transition zone, and act as structural components of the so-called “ciliary gate” to regulate the ciliary trafficking of cargo proteins or lipids. Cargo proteins include enzymes and transmembrane proteins that mediate intracellular signaling. The disruption of transition zone function may contribute to the ciliopathy phenotype by altering the composition of the ciliary membrane or axoneme, with impacts on essential developmental signaling including the Wnt and Shh pathways as well as the regulation of secondary messengers such as inositol-1,4,5-trisphosphate (InsP3) and cyclic adenosine monophosphate (cAMP). -

Evolving Concepts in Human Renal Dysplasia
DISEASE OF THE MONTH J Am Soc Nephrol 15: 998–1007, 2004 EBERHARD RITZ, FEATURE EDITOR Evolving Concepts in Human Renal Dysplasia ADRIAN S. WOOLF, KAREN L. PRICE, PETER J. SCAMBLER, and PAUL J.D. WINYARD Nephro-Urology and Molecular Medicine Units, Institute of Child Health, University College London, London, United Kingdom Abstract. Human renal dysplasia is a collection of disorders in correlating with perturbed cell turnover and maturation. Mu- which kidneys begin to form but then fail to differentiate into tations of nephrogenesis genes have been defined in multiorgan normal nephrons and collecting ducts. Dysplasia is the princi- dysmorphic disorders in which renal dysplasia can feature, pal cause of childhood end-stage renal failure. Two main including Fraser, renal cysts and diabetes, and Kallmann syn- theories have been considered in its pathogenesis: A primary dromes. Here, it is possible to begin to understand the normal failure of ureteric bud activity and a disruption produced by nephrogenic function of the wild-type proteins and understand fetal urinary flow impairment. Recent studies have docu- how mutations might cause aberrant organogenesis. mented deregulation of gene expression in human dysplasia, Congenital anomalies of the kidney and urinary tract and the main renal pathology is renal dysplasia (RD). In her (CAKUT) account for one third of all anomalies detected by landmark book Normal and Abnormal Development of the routine fetal ultrasonography (1). A recent UK audit of child- Kidney published in 1972 (7), Edith Potter emphasized that one hood end-stage renal failure reported that CAKUT was the must understand normal development to generate realistic hy- cause in ~40% of 882 individuals (2). -

Histone Deacetylase 6 Represents a Novel Drug Target in the Oncogenic Hedgehog Signaling Pathway
Aus dem Institut für Molekularbiologie und Tumorforschung Geschäftsführender Direktor: Prof. Dr. Rolf Müller des Fachbereichs Medizin der Philipps-Universität Marburg Histone deacetylase 6 represents a novel drug target in the oncogenic Hedgehog signaling pathway Inaugural- Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften dem Fachbereich Medizin der Philipps-Universität Marburg vorgelegt von Dhanyamraju Pavan Kumar aus Secunderabad, Indien Marburg-2017 Angenommen vom Fachbereich Medizin der Philipps Universität Marburg am:3/11/2017 Gedruckt mit der Genehmigung des Fachbereichs Dekan: Prof. Dr. Helmut Schäfer Referent: PD. Dr. Matthias Lauth Korreferent: Prof. Dr. Elke Pogge von Strandmann Table of Contents 1. Summary……………………………………………………………………………...1 1. Zussamenfassung …………………………………………………………....……….2 2. Introduction………………………………………………………………………......3 2.1 The Hedgehog signalling pathway…………………………………………………3 2.2 The Mechanism of Hedgehog signal transduction…………………………………5 2.3 Ptch1 a tumor suppressor gene…………………………………………………....10 2.4 Primary cilium and Hedgehog signalling.………………….………………….….11 2.5 Histone deacetylases (HDACs)………………………………………………...…15 2.6 Classification of Histone deacetylases……………………………………………16 2.7 Histone deacetylase 6……………………………………………………….….…17 2.8 Regulation of Ciliogenesis by HDAC6…………………………………………...20 2.9 Hedgehog signalling and Cancer………………………………………………….22 2.10 Hedgehog signalling Type-I- Mutation driven-ligand independent………….….22 2.11 Hedgehog signalling Type-II-Autocrine-ligand dependent………………….….23 -

Abstracts from the 51St European Society of Human Genetics Conference: Electronic Posters
European Journal of Human Genetics (2019) 27:870–1041 https://doi.org/10.1038/s41431-019-0408-3 MEETING ABSTRACTS Abstracts from the 51st European Society of Human Genetics Conference: Electronic Posters © European Society of Human Genetics 2019 June 16–19, 2018, Fiera Milano Congressi, Milan Italy Sponsorship: Publication of this supplement was sponsored by the European Society of Human Genetics. All content was reviewed and approved by the ESHG Scientific Programme Committee, which held full responsibility for the abstract selections. Disclosure Information: In order to help readers form their own judgments of potential bias in published abstracts, authors are asked to declare any competing financial interests. Contributions of up to EUR 10 000.- (Ten thousand Euros, or equivalent value in kind) per year per company are considered "Modest". Contributions above EUR 10 000.- per year are considered "Significant". 1234567890();,: 1234567890();,: E-P01 Reproductive Genetics/Prenatal Genetics then compared this data to de novo cases where research based PO studies were completed (N=57) in NY. E-P01.01 Results: MFSIQ (66.4) for familial deletions was Parent of origin in familial 22q11.2 deletions impacts full statistically lower (p = .01) than for de novo deletions scale intelligence quotient scores (N=399, MFSIQ=76.2). MFSIQ for children with mater- nally inherited deletions (63.7) was statistically lower D. E. McGinn1,2, M. Unolt3,4, T. B. Crowley1, B. S. Emanuel1,5, (p = .03) than for paternally inherited deletions (72.0). As E. H. Zackai1,5, E. Moss1, B. Morrow6, B. Nowakowska7,J. compared with the NY cohort where the MFSIQ for Vermeesch8, A. -

Irish Rare Kidney Disease Network (IRKDN)
Irish Rare kidney Disease Network (IRKDN) Others Cork University Mater, Waterford University Dr Liam Plant Hospital Galway Dr Abernathy University Hospital Renal imaging Dr M Morrin Prof Griffin Temple St and Crumlin Beaumont Hospital CHILDRENS Hospital Tallaght St Vincents Dr Atiff Awann Rare Kidney Disease Clinic Hospital University Hospital Prof Peter Conlon Dr Lavin Prof Dr Holian Little Renal pathology Lab Limerick University Dr Dorman and Hospital Dr Doyle Dr Casserly Patient Renal Council Genetics St James Laboratory Hospital RCSI Dr Griffin Prof Cavaller MISION Provision of care to patients with Rare Kidney Disease based on best available medical evidence through collaboration within Ireland and Europe Making available clinical trials for rare kidney disease to Irish patients where available Collaboration with other centres in Europe treating rare kidney disease Education of Irish nephrologists on rare Kidney Disease. Ensuring a seamless transition of children from children’s hospital with rare kidney disease to adult centres with sharing of knowledge of rare paediatric kidney disease with adult centres The provision of precise molecular diagnosis of patients with rare kidney disease The provision of therapeutic plan based on understanding of molecular diagnosis where available Development of rare disease specific registries within national renal It platform ( Emed) Structure Beaumont Hospital will act as National rare Kidney Disease Coordinating centre working in conjunction with a network of Renal unit across the country -

Meckel–Gruber Syndrome: an Update on Diagnosis, Clinical Management, and Research Advances
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by White Rose Research Online MINI REVIEW published: 20 November 2017 doi: 10.3389/fped.2017.00244 Meckel–Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances Verity Hartill1,2, Katarzyna Szymanska2, Saghira Malik Sharif1, Gabrielle Wheway3 and Colin A. Johnson2* 1 Department of Clinical Genetics, Yorkshire Regional Genetics Service, Leeds Teaching Hospitals NHS Trust, Leeds, United Kingdom, 2 Leeds Institute of Biomedical and Clinical Sciences, University of Leeds, Leeds, United Kingdom, 3 Faculty of Health and Applied Sciences, Department of Applied Sciences, UWE Bristol, Bristol, United Kingdom Meckel–Gruber syndrome (MKS) is a lethal autosomal recessive congenital anomaly syndrome caused by mutations in genes encoding proteins that are structural or func- tional components of the primary cilium. Conditions that are caused by mutations in ciliary genes are collectively termed the ciliopathies, and MKS represents the most severe condition in this group of disorders. The primary cilium is a microtubule-based organelle, projecting from the apical surface of vertebrate cells. It acts as an “antenna” Edited by: that receives and transduces chemosensory and mechanosensory signals, but also Miriam Schmidts, regulates diverse signaling pathways, such as Wnt and Shh, that have important roles Radboud University Nijmegen, Netherlands during embryonic development. Most MKS proteins localize to a distinct ciliary com- Reviewed by: partment called the transition zone (TZ) that regulates the trafficking of cargo proteins Julia Hoefele, or lipids. In this review, we provide an up-to-date summary of MKS clinical features, Technische Universität München, Germany molecular genetics, and clinical diagnosis. -
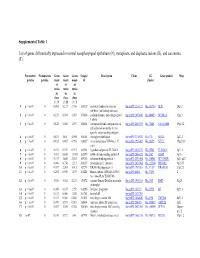
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

Ciliopathies
T h e new england journal o f medicine Review article Mechanisms of Disease Robert S. Schwartz, M.D., Editor Ciliopathies Friedhelm Hildebrandt, M.D., Thomas Benzing, M.D., and Nicholas Katsanis, Ph.D. iverse developmental and degenerative single-gene disor- From the Howard Hughes Medical Insti- ders such as polycystic kidney disease, nephronophthisis, retinitis pigmen- tute and the Departments of Pediatrics and Human Genetics, University of Michi- tosa, the Bardet–Biedl syndrome, the Joubert syndrome, and the Meckel gan Health System, Ann Arbor (F.H.); the D Renal Division, Department of Medicine, syndrome may be categorized as ciliopathies — a recent concept that describes dis- eases characterized by dysfunction of a hairlike cellular organelle called the cilium. Center for Molecular Medicine, and Co- logne Cluster of Excellence in Cellular Most of the proteins that are altered in these single-gene disorders function at the Stress Responses in Aging-Associated Dis- level of the cilium–centrosome complex, which represents nature’s universal system eases, University of Cologne, Cologne, for cellular detection and management of external signals. Cilia are microtubule- Germany (T.B.); and the Center for Hu- man Disease Modeling and the Depart- based structures found on almost all vertebrate cells. They originate from a basal ments of Pediatrics and Cell Biology, body, a modified centrosome, which is the organelle that forms the spindle poles Duke University Medical Center, Durham, during mitosis. The important role that the cilium–centrosome complex plays in NC (N.K.). Address reprint requests to Dr. Hildebrandt at Howard Hughes Med- the normal function of most tissues appears to account for the involvement of mul- ical Institute, Departments of Pediatrics tiple organ systems in ciliopathies. -

Ciliary Genes in Renal Cystic Diseases
cells Review Ciliary Genes in Renal Cystic Diseases Anna Adamiok-Ostrowska * and Agnieszka Piekiełko-Witkowska * Department of Biochemistry and Molecular Biology, Centre of Postgraduate Medical Education, 01-813 Warsaw, Poland * Correspondence: [email protected] (A.A.-O.); [email protected] (A.P.-W.); Tel.: +48-22-569-3810 (A.P.-W.) Received: 3 March 2020; Accepted: 5 April 2020; Published: 8 April 2020 Abstract: Cilia are microtubule-based organelles, protruding from the apical cell surface and anchoring to the cytoskeleton. Primary (nonmotile) cilia of the kidney act as mechanosensors of nephron cells, responding to fluid movements by triggering signal transduction. The impaired functioning of primary cilia leads to formation of cysts which in turn contribute to development of diverse renal diseases, including kidney ciliopathies and renal cancer. Here, we review current knowledge on the role of ciliary genes in kidney ciliopathies and renal cell carcinoma (RCC). Special focus is given on the impact of mutations and altered expression of ciliary genes (e.g., encoding polycystins, nephrocystins, Bardet-Biedl syndrome (BBS) proteins, ALS1, Oral-facial-digital syndrome 1 (OFD1) and others) in polycystic kidney disease and nephronophthisis, as well as rare genetic disorders, including syndromes of Joubert, Meckel-Gruber, Bardet-Biedl, Senior-Loken, Alström, Orofaciodigital syndrome type I and cranioectodermal dysplasia. We also show that RCC and classic kidney ciliopathies share commonly disturbed genes affecting cilia function, including VHL (von Hippel-Lindau tumor suppressor), PKD1 (polycystin 1, transient receptor potential channel interacting) and PKD2 (polycystin 2, transient receptor potential cation channel). Finally, we discuss the significance of ciliary genes as diagnostic and prognostic markers, as well as therapeutic targets in ciliopathies and cancer. -

Prenatal Versus Postnatal Diagnosis of Meckel–Gruber and Joubert Syndrome in Patients with TMEM67 Mutations
G C A T T A C G G C A T genes Article Prenatal Versus Postnatal Diagnosis of Meckel–Gruber and Joubert Syndrome in Patients with TMEM67 Mutations Agnieszka Stembalska 1,* , Małgorzata Rydzanicz 2 , Agnieszka Pollak 2, Grazyna Kostrzewa 2, Piotr Stawinski 2, Mateusz Biela 3 , Rafal Ploski 2 and Robert Smigiel 3,* 1 Department of Genetics, Wroclaw Medical University, 50-368 Wroclaw, Poland 2 Department of Medical Genetics, Medical University of Warsaw, 02-106 Warsaw, Poland; [email protected] (M.R.); [email protected] (A.P.); [email protected] (G.K.); [email protected] (P.S.); [email protected] (R.P.) 3 Department of Paediatrics, Division of Paediatric Propedeutics and Rare Disorders, Wroclaw Medical University, 51-618 Wroclaw, Poland; [email protected] * Correspondence: [email protected] (A.S.); [email protected] (R.S.) Abstract: Renal cystic diseases are characterized by genetic and phenotypic heterogeneity. Congenital renal cysts can be classified as developmental disorders and are commonly diagnosed prenatally using ultrasonography and magnetic resonance imaging. Progress in molecular diagnostics and availability of exome sequencing procedures allows diagnosis of single-gene disorders in the prenatal period. Two patients with a prenatal diagnosis of polycystic kidney disease are presented in this article. TMEM67 mutations were identified in both fetuses using a whole-exome sequencing (WES) study. In one of them, the phenotypic syndrome diagnosed prenatally was different from that Citation: Stembalska, A.; Rydzanicz, diagnosed in the postnatal period. M.; Pollak, A.; Kostrzewa, G.; Stawinski, P.; Biela, M.; Ploski, R.; Keywords: TMEM67 gene; prenatal and postnatal diagnosis; genetic and phenotypic diagnosis; Smigiel, R. -

Department of Pathology and Laboratory Medicine
University of Kansas Medical Center Department of Pathology and Laboratory Medicine Resident Manual (2012-2013) Table of Contents Mission, Goals and Philosophy ........................................................................................................4 Educational Program........................................................................................................................5 Overall Educational Goals ..............................................................................................................5 Program Structure .........................................................................................................................10 PGY-Specific Goals ......................................................................................................................12 Didactic Sessions and Conferences ..............................................................................................17 Resident Scholarly Activity ..........................................................................................................17 USMLE Step 3 Policy ...................................................................................................................18 Professionalism .............................................................................................................................19 Eligibility and Selection of Residents ...........................................................................................20 Evaluations ....................................................................................................................................21