Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus Bawanglingensis Huang, Li Et Xing, a Vulnerable Oak Tree in China
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

5 Fagaceae Trees
CHAPTER 5 5 Fagaceae Trees Antoine Kremerl, Manuela Casasoli2,Teresa ~arreneche~,Catherine Bod6n2s1, Paul Sisco4,Thomas ~ubisiak~,Marta Scalfi6, Stefano Leonardi6,Erica ~akker~,Joukje ~uiteveld', Jeanne ~omero-Seversong, Kathiravetpillai Arumuganathanlo, Jeremy ~eror~',Caroline scotti-~aintagne", Guy Roussell, Maria Evangelista Bertocchil, Christian kxerl2,Ilga porth13, Fred ~ebard'~,Catherine clark15, John carlson16, Christophe Plomionl, Hans-Peter Koelewijn8, and Fiorella villani17 UMR Biodiversiti Genes & Communautis, INRA, 69 Route d'Arcachon, 33612 Cestas, France, e-mail: [email protected] Dipartimento di Biologia Vegetale, Universita "La Sapienza", Piazza A. Moro 5,00185 Rome, Italy Unite de Recherche sur les Especes Fruitikres et la Vigne, INRA, 71 Avenue Edouard Bourlaux, 33883 Villenave d'Ornon, France The American Chestnut Foundation, One Oak Plaza, Suite 308 Asheville, NC 28801, USA Southern Institute of Forest Genetics, USDA-Forest Service, 23332 Highway 67, Saucier, MS 39574-9344, USA Dipartimento di Scienze Ambientali, Universitk di Parma, Parco Area delle Scienze 1lIA, 43100 Parma, Italy Department of Ecology and Evolution, University of Chicago, 5801 South Ellis Avenue, Chicago, IL 60637, USA Alterra Wageningen UR, Centre for Ecosystem Studies, P.O. Box 47,6700 AA Wageningen, The Netherlands Department of Biological Sciences, University of Notre Dame, Notre Dame, IN 46556, USA lo Flow Cytometry and Imaging Core Laboratory, Benaroya Research Institute at Virginia Mason, 1201 Ninth Avenue, Seattle, WA 98101, -
Qrno. 1 2 3 4 5 6 7 1 CP 2903 77 100 0 Cfcl3
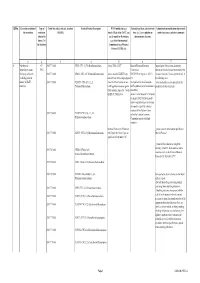
QRNo. General description of Type of Tariff line code(s) affected, based on Detailed Product Description WTO Justification (e.g. National legal basis and entry into Administration, modification of previously the restriction restriction HS(2012) Article XX(g) of the GATT, etc.) force (i.e. Law, regulation or notified measures, and other comments (Symbol in and Grounds for Restriction, administrative decision) Annex 2 of e.g., Other International the Decision) Commitments (e.g. Montreal Protocol, CITES, etc) 12 3 4 5 6 7 1 Prohibition to CP 2903 77 100 0 CFCl3 (CFC-11) Trichlorofluoromethane Article XX(h) GATT Board of Eurasian Economic Import/export of these ozone destroying import/export ozone CP-X Commission substances from/to the customs territory of the destroying substances 2903 77 200 0 CF2Cl2 (CFC-12) Dichlorodifluoromethane Article 46 of the EAEU Treaty DECISION on August 16, 2012 N Eurasian Economic Union is permitted only in (excluding goods in dated 29 may 2014 and paragraphs 134 the following cases: transit) (all EAEU 2903 77 300 0 C2F3Cl3 (CFC-113) 1,1,2- 4 and 37 of the Protocol on non- On legal acts in the field of non- _to be used solely as a raw material for the countries) Trichlorotrifluoroethane tariff regulation measures against tariff regulation (as last amended at 2 production of other chemicals; third countries Annex No. 7 to the June 2016) EAEU of 29 May 2014 Annex 1 to the Decision N 134 dated 16 August 2012 Unit list of goods subject to prohibitions or restrictions on import or export by countries- members of the -

New York City Approved Street Trees
New York City Approved Street Trees Suggested Tree Species Shape Visual interest Frequency of Preferred Cultivars Notes Scientific Name Common Name Planting Acer rubrum Red Maple Sparingly 'Red Sunset' ALB Host Aesculus hippocastanum Horsechestnut White May flowers Sparingly 'Baumanni' ALB Host Aesculus octandra Yellow Buckeye Yellow May Flowers Sparingly ALB Host ALB Host 'Duraheat' Betula nigra River Birch Ornamental Bark Sparingly Plant Single Stem 'Heritage' Only Celtis occidentalis Hackberry Ornamental Bark Sparingly 'Magnifica' ALB Host ALB Host Cercidiphyllum japonicum Katsura Tree Sparingly Plant Single Stem Only Corylus colurna Turkish Filbert Sparingly LARGE TREES: Mature LARGE TREES: height than greater feet 50 tall Eucommia ulmoides Hardy Rubber Tree Frequently 'Asplenifolia' Fagus sylvatica European Beech Sparingly 'Dawyckii Purple' 'Autumn Gold' Ginkgo biloba Ginkgo Yellow Fall Color Moderately 'Magyar' Very Tough Tree 'Princeton Sentry' 'Shademaster' 'Halka' Gleditsia triacanthos var inermis Honeylocust Yellow Fall Color Moderately 'Imperial' 'Skyline' 'Espresso' Gymnocladus dioicus Kentucky Coffeetree Large Tropical Leaves Frequently 'Prairie Titan' Page 1 of 7 New York City Approved Street Trees Suggested Tree Species Shape Visual interest Frequency of Preferred Cultivars Notes Scientific Name Common Name Planting 'Rotundiloba' Seedless Cultivars Liquidambar styraciflua Sweetgum Excellent Fall Color Frequently 'Worplesdon' Preffered 'Cherokee' Orange/Green June Liriodendron tulipifera Tulip Tree Moderately Flowers Metasequoia -

Perennials 2016
Shady Acres Perennial Nursery 1958 - 2016 New Berlin, WI years58 in the Green Industry Perennials 2016 www.ShadyAcresNursery.com 262-679-1610 e-mail: [email protected] Planting Guide Perennial Border Shade r Sun e d Perennials r o Perennials B l a i n Hosta n e r e Ferns P Woodlands Groundcovers Store Cashier Clematis Grasses Shade Garden Shade Herbs Sunny Garden & Offices Shade Annuals More Shade Gazebo Create-a- Potting Soil Container Cocoa Bean Mulch Entrance Mushroom Compost Cotton Burr ~ Parking Area ~ Compost Pine Needle 0 Mulch Garbe Enterprises, Inc. dba Shady Acres Perennial Nursery 5725 South Martin Road New Berlin, Wisconsin 53146-5112 phone: 262-679-1610 e-mail: [email protected] web: www.ShadyAcresNursery.com Welcome to Shady Acres Perennial Nursery ~ 2016 Gardening Season ~ Shady Acres is your destination for locally grown perennials. Since 1958, our family-owned business has been serving the green industry. Perennials are our specialty, and we are known for our outstanding selection of top-quality plants. We offer over 1200 varieties including perennials for sun or shade, ornamental grasses, native plants, groundcovers and fresh herbs! We invite you to browse our garden center, where all the plants we grow are arranged in an easy-to-find fashion. Perennials are the perfect choice for enhancing your outdoor areas. A garden that changes through all four seasons will bring years of beauty and enjoyment. Stroll through our many display gardens to see mature perennials growing in a garden environment. Come often to observe how the foliage and flowers change throughout the season. -

The First Chloroplast Genome Sequence of Boswellia Sacra, a Resin-Producing Plant in Oman
RESEARCH ARTICLE The First Chloroplast Genome Sequence of Boswellia sacra, a Resin-Producing Plant in Oman Abdul Latif Khan1, Ahmed Al-Harrasi1*, Sajjad Asaf2, Chang Eon Park2, Gun-Seok Park2, Abdur Rahim Khan2, In-Jung Lee2, Ahmed Al-Rawahi1, Jae-Ho Shin2* 1 UoN Chair of Oman's Medicinal Plants & Marine Natural Products, University of Nizwa, Nizwa, Oman, 2 School of Applied Biosciences, Kyungpook National University, Daegu, Republic of Korea a1111111111 * [email protected] (AAH); [email protected] (JHS) a1111111111 a1111111111 a1111111111 Abstract a1111111111 Boswellia sacra (Burseraceae), a keystone endemic species, is famous for the production of fragrant oleo-gum resin. However, the genetic make-up especially the genomic informa- tion about chloroplast is still unknown. Here, we described for the first time the chloroplast OPEN ACCESS (cp) genome of B. sacra. The complete cp sequence revealed a circular genome of 160,543 Citation: Khan AL, Al-Harrasi A, Asaf S, Park CE, bp size with 37.61% GC content. The cp genome is a typical quadripartite chloroplast struc- Park G-S, Khan AR, et al. (2017) The First ture with inverted repeats (IRs 26,763 bp) separated by small single copy (SSC; 18,962 bp) Chloroplast Genome Sequence of Boswellia sacra, and large single copy (LSC; 88,055 bp) regions. De novo assembly and annotation showed a Resin-Producing Plant in Oman. PLoS ONE 12 the presence of 114 unique genes with 83 protein-coding regions. The phylogenetic analysis (1): e0169794. doi:10.1371/journal.pone.0169794 revealed that the B. sacra cp genome is closely related to the cp genome of Azadirachta Editor: Xiu-Qing Li, Agriculture and Agri-Food indica and Citrus sinensis, while most of the syntenic differences were found in the non-cod- Canada, CANADA ing regions. -

(Castanea) Cultivar 'Yanshanzaofeng'
Advance Journal of Food Science and Technology 5(9): 1192-1197, 2013 ISSN: 2042-4868; e-ISSN: 2042-4876 © Maxwell Scientific Organization, 2013 Submitted: May 20, 2013 Accepted: June 11, 2013 Published: September 05, 2013 A Morphological and Histological Characterization of Male Flower in Chestnut (Castanea) Cultivar ‘Yanshanzaofeng’ Feng Zou, Su-Juan Guo, Huan Xiong, Peng Xie, Wen-Jun Lv and Guang-Hui Li Key Laboratory for Silviculture and Conservation, Ministry of Education, Beijing Forestry University, 100083, Beijing, P.R. China Abstract: Chinese chestnut (Castanea mollissima Blume.) is a widely distributed fruit tree and well known for its ecological and economic value. In order to evaluate obstacles to male reproductive in the C. mollissima, a morphological and histological characterization of male flower of chestnut cultivar ‘Yanshanzaofeng’ were examined by paraffin section technique and scanning electron microscopy. The results showed that male catkins with floral primordia were formed in the buds of one-year olds shoots in later April. Later, a protoderm, ground meristem and a procambium had differentiated in young anthers. Each young anther soon developed to four microsporangia. The anther wall layers developed completely by mid-May and consisted of one-cell-layered epidermis, one-cell-layered endothecium, two or three middle layers and one-cell-layered tapetum. The tapetum was of glandular type. Microspore mother cells underwent meiosis through simultaneous cytokinesis in later May and gave rise to tetrads of microspores, which were tetrahedrally arranged. Mature pollens contained two cells with three germ pores. Anthers were dehiscent and pollen grains shed by early June. Based our results, we did not find the abnormal male flower in the C. -

Chapter 4 Phytogeography of Northeast Asia
Chapter 4 Phytogeography of Northeast Asia Hong QIAN 1, Pavel KRESTOV 2, Pei-Yun FU 3, Qing-Li WANG 3, Jong-Suk SONG 4 and Christine CHOURMOUZIS 5 1 Research and Collections Center, Illinois State Museum, 1011 East Ash Street, Springfield, IL 62703, USA, e-mail: [email protected]; 2 Institute of Biology and Soil Science, Russian Academy of Sciences, Vladivostok, 690022, Russia, e-mail: [email protected]; 3 Institute of Applied Ecology, Chinese Academy of Sciences, P.O. Box 417, Shenyang 110015, China; 4 Department of Biological Science, College of Natural Sciences, Andong National University, Andong 760-749, Korea, e-mail: [email protected]; 5 Department of Forest Sciences, University of British Columbia, 3041-2424 mail Mall, Vancouver, B.C., V6T 1Z4, Canada, e-mail: [email protected] Abstract: Northeast Asia as defined in this study includes the Russian Far East, Northeast China, the northern part of the Korean Peninsula, and Hokkaido Island (Japan). We determined the species richness of Northeast Asia at various spatial scales, analyzed the floristic relationships among geographic regions within Northeast Asia, and compared the flora of Northeast Asia with surrounding floras. The flora of Northeast Asia consists of 971 genera and 4953 species of native vascular plants. Based on their worldwide distributions, the 971 gen- era were grouped into fourteen phytogeographic elements. Over 900 species of vascular plants are endemic to Northeast Asia. Northeast Asia shares 39% of its species with eastern Siberia-Mongolia, 24% with Europe, 16.2% with western North America, and 12.4% with eastern North America. -

Quercus Drymeja Unger and Q. Mediterranea Unger
Review of Palaeobotany and Palynology 241 (2017) 98–128 Contents lists available at ScienceDirect Review of Palaeobotany and Palynology journal homepage: www.elsevier.com/locate/revpalbo Taxonomy and palaeoecology of two widespread western Eurasian Neogene sclerophyllous oak species: Quercus drymeja Unger and Q. mediterranea Unger Thomas Denk a,⁎, Dimitrios Velitzelos b,TuncayH.Günerc, Johannes M. Bouchal a,d, Friðgeir Grímsson d,GuidoW.Grimmd,e a Swedish Museum of Natural History, Department of Palaeobiology, Box 50007, 10405 Stockholm, Sweden b National and Kapodistrian University of Athens, Faculty of Geology and Geoenvironment, Department of Historical Geology and Paleontology, Panepistimiopolis, Athens 15784, Greece c Istanbul University, Faculty of Forestry, Department of Forest Botany, 34473 Bahceköy, Istanbul, Turkey d University of Vienna, Department of Palaeontology, 1090 Vienna, Austria e Unaffiliated, 45100 Orléans, France article info abstract Article history: Sclerophyllous oaks (genus Quercus) play important roles in Neogene ecosystems of south-western Eurasia. Received 31 May 2016 Modern analogues (‘nearest living relatives’) for these oaks have been sought among five of six infrageneric lin- Accepted 30 January 2017 eages of Quercus, distributed across the entire Northern Hemisphere. A revision of leaf fossils from lower Miocene Available online 10 February 2017 to Pliocene deposits suggests that morphotypes of the Quercus drymeja complex are very similar to a number of extant Himalayan, East Asian, and Southeast Asian species of Quercus Group Ilex and may indicate subtropical, Keywords: Quercus Group Ilex relatively humid conditions. Quercus mediterranea comprises leaf morphotypes that are encountered in modern Plant fossil Mediterranean species of Quercus Group Ilex, but also in Himalayan and East Asian members of this group indi- Modern analogue cating fully humid or summer-wet conditions. -

Saxifragaceae
Flora of China 8: 269–452. 2001. SAXIFRAGACEAE 虎耳草科 hu er cao ke Pan Jintang (潘锦堂)1, Gu Cuizhi (谷粹芝 Ku Tsue-chih)2, Huang Shumei (黄淑美 Hwang Shu-mei)3, Wei Zhaofen (卫兆芬 Wei Chao-fen)4, Jin Shuying (靳淑英)5, Lu Lingdi (陆玲娣 Lu Ling-ti)6; Shinobu Akiyama7, Crinan Alexander8, Bruce Bartholomew9, James Cullen10, Richard J. Gornall11, Ulla-Maj Hultgård12, Hideaki Ohba13, Douglas E. Soltis14 Herbs or shrubs, rarely trees or vines. Leaves simple or compound, usually alternate or opposite, usually exstipulate. Flowers usually in cymes, panicles, or racemes, rarely solitary, usually bisexual, rarely unisexual, hypogynous or ± epigynous, rarely perigynous, usually biperianthial, rarely monochlamydeous, actinomorphic, rarely zygomorphic, 4- or 5(–10)-merous. Sepals sometimes petal-like. Petals usually free, sometimes absent. Stamens (4 or)5–10 or many; filaments free; anthers 2-loculed; staminodes often present. Carpels 2, rarely 3–5(–10), usually ± connate; ovary superior or semi-inferior to inferior, 2- or 3–5(–10)-loculed with axile placentation, or 1-loculed with parietal placentation, rarely with apical placentation; ovules usually many, 2- to many seriate, crassinucellate or tenuinucellate, sometimes with transitional forms; integument 1- or 2-seriate; styles free or ± connate. Fruit a capsule or berry, rarely a follicle or drupe. Seeds albuminous, rarely not so; albumen of cellular type, rarely of nuclear type; embryo small. About 80 genera and 1200 species: worldwide; 29 genera (two endemic), and 545 species (354 endemic, seven introduced) in China. During the past several years, cladistic analyses of morphological, chemical, and DNA data have made it clear that the recognition of the Saxifragaceae sensu lato (Engler, Nat. -

Karyotypic Relationships Among Astilboides, Bergenia, Darmera, and Mukdenia and Their Implications for Subtribal Boundaries in Saxifrageae (Saxifragaceae)
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/237162727 Karyotypic relationships among Astilboides, Bergenia, Darmera, and Mukdenia and their implications for subtribal boundaries in Saxifrageae (Saxifragaceae) Article in Canadian Journal of Botany · January 2011 DOI: 10.1139/b86-075 CITATIONS READS 3 72 1 author: Douglas E Soltis University of Florida 1,009 PUBLICATIONS 76,596 CITATIONS SEE PROFILE Some of the authors of this publication are also working on these related projects: Systematics and evolution of Amaryllidaceae tribe Hippeastreae View project Teaching Plant Systematics View project All content following this page was uploaded by Douglas E Soltis on 01 October 2014. The user has requested enhancement of the downloaded file. Karyotypic relationships among Astilboides, Bergenia, Darmera, and Mukdenia and their implications for subtribal boundaries in Saxifrageae (Saxifragaceae) Department of Botany, Washington State Universiv, Pullman, WA, U.S.A. 99164-4330 Received May 7, 1985 SOLTIS,D. E. 1986. Karyotypic relationships among Astilboides, Bergenia, Dar~nera,and Mukdenia and their implications for subtribal boundaries in Saxifrageae (Saxifragaceae). Can. J. Bot. 64: 586-588. Chromosomal studies have revealed that 2n = 34 in Astilboides, Darmera, Mukdenia, and Bergenia and that these genera have very similar karyotypes. Few taxa of Saxifrageae have x = 17, and chromosomal data therefore suggest that these four genera form a natural group. The generic affinities suggested by chromosomal data are not in agreement with the traditional interpretation of relationships in Saxifrageae. Astilboides has been placed in subtribe Astilbinae, while Bergenia, Darmera, and Mukdenia were considered members of Saxifraginae. The data presented here, in conjunction with data from earlier chromosomal studies, indicate that traditional subtribal boundaries do not accurately reflect relationships in tribe Saxifrageae. -

Download the New York Botanical Garden Collections Master Plan Quercus.Pdf
The New York Botanical Garden Collections Master Plan Quercus Quercus Peter Del Tredici, Paul Meyer, Todd Forrest, Jessica Arcate I. Introduction Mature oaks, both planted and preexisting, dominate The New York Botanical Garden’s landscape. A variety of native species grow throughout the Botanical Garden’s grounds and Forest. Many of these existing specimens are over 200 years old. The Oak Collection, located along Magnolia Way, is one of three intact systematic collections that remain from the original Deciduous Arboretum established in 1896. Here mature native species, exotic species, and hybrids grow side by side. The Garden’s oak collection is aging. Very few middle-aged specimens and almost no young specimens are present. This presents a problem if the Garden hopes to maintain the abundance of mature oaks that create the distinctive character of the landscape. The loss of some of the oldest mature oaks in the Forest in recent droughts and severe storms underscores the importance of maintaining a wide range of species and age classes in the living collections. Since oaks are so widespread at the Garden, new specimens could be planted on any appropriate site across the Garden’s 250-acre landscape. II. Analysis A. Summarize taxonomic diversity of Collection or Garden The Garden’s oak collection includes primarily regionally native species and hybrids. Only a fraction of the European, Asian, and other North American taxa hardy in our climate are present. B. List noteworthy taxa and specimens Noteworthy introduced oaks in the Garden’s living collections include a mature Quercus dentata at the Mosholu Gate, a massive Quercus cerris in the Oak Collection, and a large Quercus acutissima , also in the Oak Collection. -

FAGACEAE 1. FAGUS Linnaeus, Sp. Pl. 2: 997. 1753
Flora of China 4: 314–400. 1999. 1 FAGACEAE 壳斗科 qiao dou ke Huang Chengjiu (黄成就 Huang Ching-chieu)1, Zhang Yongtian (张永田 Chang Yong-tian)2; Bruce Bartholomew3 Trees or rarely shrubs, monoecions, evergreen or deciduous. Stipules usually early deciduous. Leaves alternate, sometimes false-whorled in Cyclobalanopsis. Inflorescences unisexual or androgynous with female cupules at the base of an otherwise male inflorescence. Male inflorescences a pendulous head or erect or pendulous catkin, sometimes branched; flowers in dense cymules. Male flower: sepals 4–6(–9), scalelike, connate or distinct; petals absent; filaments filiform; anthers dorsifixed or versatile, opening by longitudinal slits; with or without a rudimentary pistil. Female inflorescences of 1–7 or more flowers subtended individually or collectively by a cupule formed from numerous fused bracts, arranged individually or in small groups along an axis or at base of an androgynous inflorescence or on a separate axis. Female flower: perianth 1–7 or more; pistil 1; ovary inferior, 3–6(– 9)-loculed; style and carpels as many as locules; placentation axile; ovules 2 per locule. Fruit a nut. Seed usually solitary by abortion (but may be more than 1 in Castanea, Castanopsis, Fagus, and Formanodendron), without endosperm; embryo large. Seven to 12 genera (depending on interpretation) and 900–1000 species: worldwide except for tropical and S Africa; seven genera and 294 species (163 endemic, at least three introduced) in China. Many species are important timber trees. Nuts of Fagus, Castanea, and of most Castanopsis species are edible, and oil is extracted from nuts of Fagus. Nuts of most species of this family contain copious amounts of water soluble tannin.